IGV and visualisation
Learning outcomes
After having completed this chapter you will be able to:
- Prepare a bam file for loading it into IGV
- Use IGV to:
- Navigate through a reference genome and alignments
- Retrieve information on a specific alignment
- Investigate (possible) variants
- Identify repeats and large INDELs
Material
The exercises below are partly based on this tutorial from the Griffith lab.
Exercises
A first glance: the E. coli dataset
Index the alignment that was filtered for the region between 2000 and 2500 kb:
cd ~/workdir/alignment_output
samtools index SRR519926.sorted.region.bam
SRR519926.sorted.region.bam.bai) and the reference genome (ecoli-strK12-MG1655.fasta) to your desktop.
If working with Docker
If you are working with Docker, you can find the files in the working directory that you mounted to the docker container (with the -v option). So if you have used -v C:\Users\myusername\ngs-course:/root/workdir, your files will be in C:\Users\myusername\ngs-course.
- Load the genome (
.fasta) into IGV: Genomes > Load Genome from File… - Load the alignment file (
.bam): File > Load from File… -
Zoom in into the region U00096.3:2046000-2048000. You can do this in two ways:
- With the search box
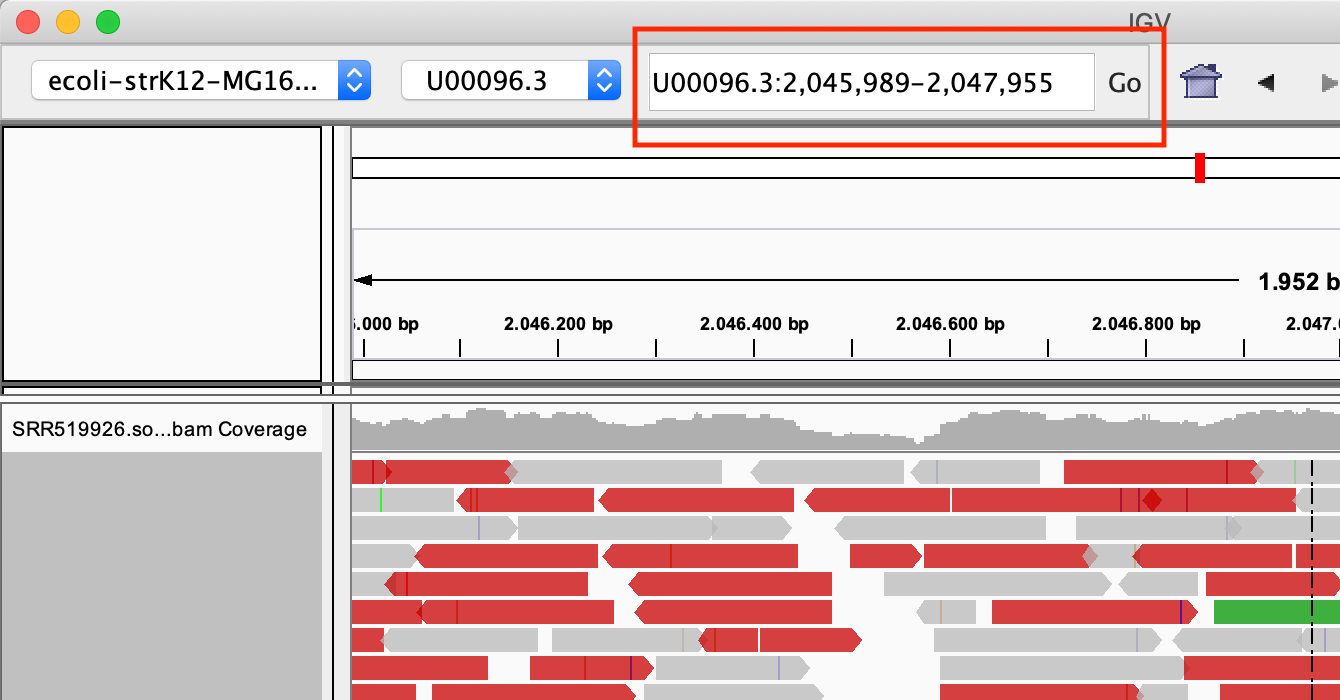
- Select the region in the location bar

-
View the reads as pairs, by right click on the reads and select View as pairs
Exercise: There are lot of reads that are coloured red. Why is that?
Answer
According to IGV, reads are coloured red if the insert size is larger than expected. As you remember, this dataset has a very large variation in insert size.
Modify the popup text behaviour by clicking on the yellow balloon to Show Details on Click:
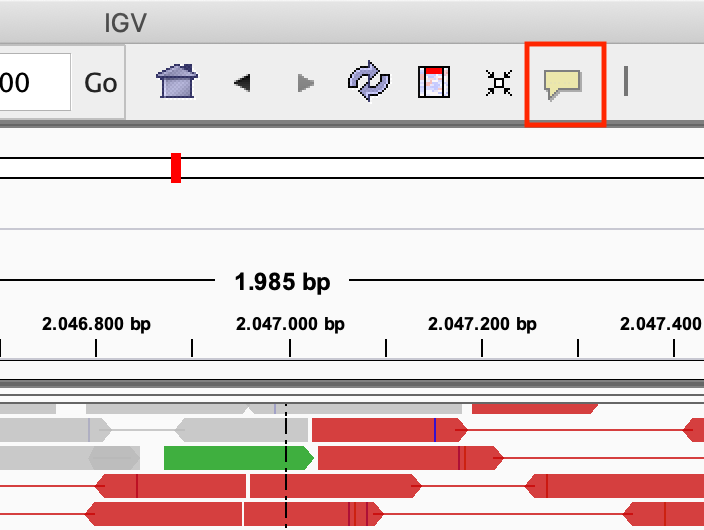
Exercise: Click on one of the reads. What kind of information is there?
Answer
Most of the information from the SAM file.
Colour the alignment by pair orientation by right clicking on the reads, and click Color alignments by > read strand.
HCC1143 data set
For this part, we will be using publicly available Illumina sequence data generated for the HCC1143 cell line. The HCC1143 cell line was generated from a 52 year old caucasian woman with breast cancer.
Sequence reads were aligned to version GRCh37 of the human reference genome. We will be working with subsets of aligned reads in the region: chromosome 21: 19,000,000 - 20,000,000.
The BAM files containing these reads for the cancer cell line and the matched normal are:
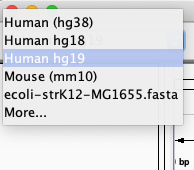
A lot of model-organism genomes are built-in IGV. Select the human genome version hg19 from the drop down menu:
Select File > Load from File… from the main menu and select the BAM file HCC1143.normal.21.19M-20M.bam using the file browser.
This BAM file only contains data for a 1 Megabase region of chromosome 21. Let’s navigate there to see what genes this region covers. To do so, navigate to chr21:19,000,000-20,000,000.
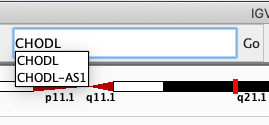
Navigate to the gene CHODL by typing it in the search box.
Load the dbsnp annotations by clicking File > Load From Server… > Annotations > Variation and Repeats > dbSNP 1.4.7
Like you did with the gene (i.e. by typing it in the search box), navigate to SNP rs3827160 that is annotated in the loaded file.
Click on the coverage track where the SNP is:
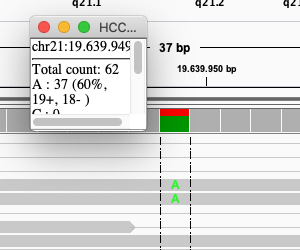
Exercise: What is the sequence coverage for that base? And the percentage T?
Answer
The coverage is 62, and 25 reads (40%) T.
Navigate to region chr21:19,800,320-19,818,162
Load repeat tracks by selecting File > Load from Server… from the main menu and then select Annotations > Variation and Repeats > Repeat Masker
Note
This might take a while to load.
Right click in the alignment track and select Color alignments by > insert size and pair orientation
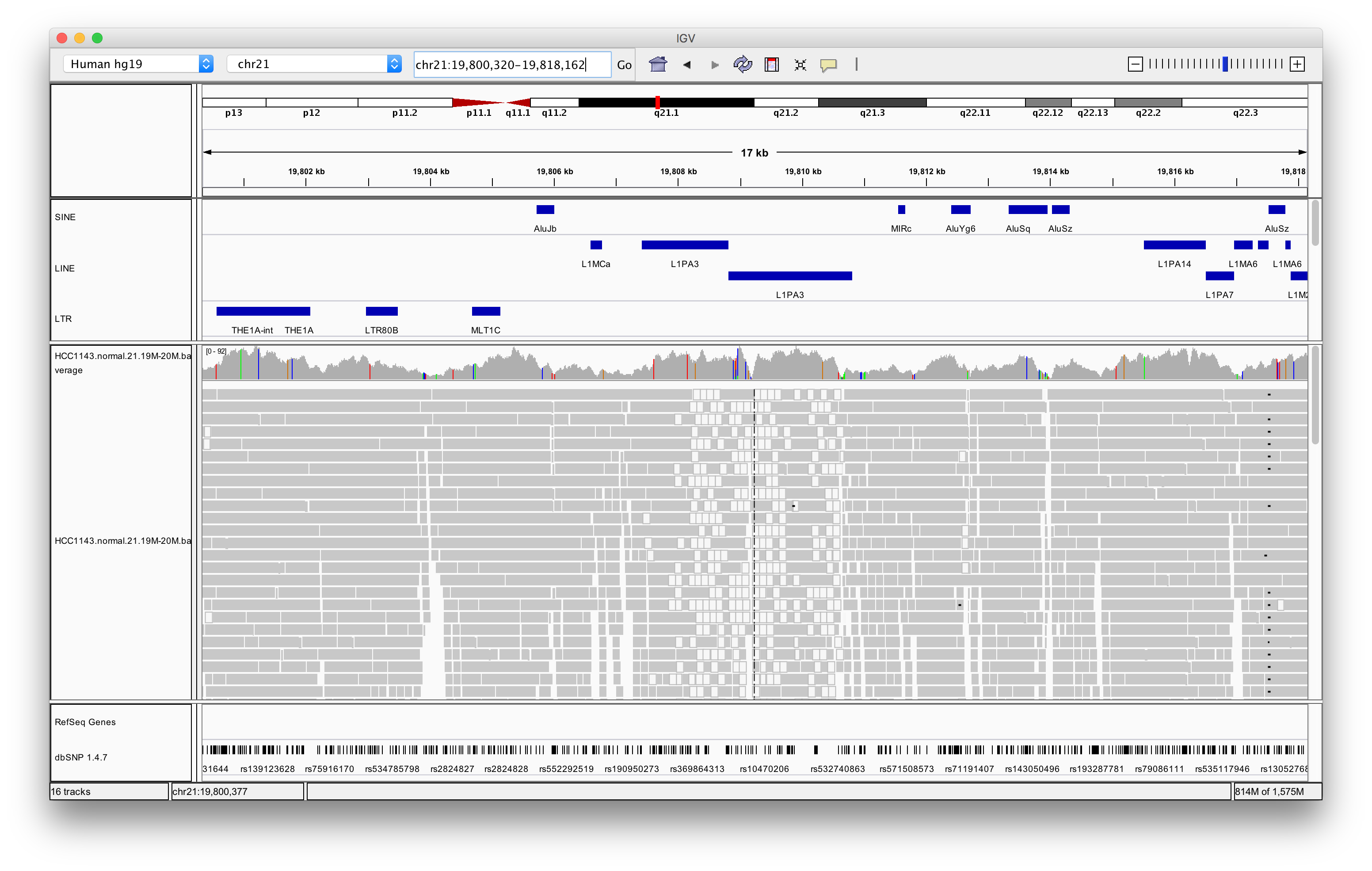
Exercise: Why are some reads coloured white? What can be the cause of that?
Answer
The white coloured reads have a map quality of 0 (click on the read to find the mapping quality). The cause of that is a LINE repeat region called L1PA3.
Navigate to region chr21:19,324,500-19,331,500
Right click in the main alignment track and select:
- Expanded
- View as pairs
- Color alignments by > insert size and pair orientation
- Sort alignments by > insert size
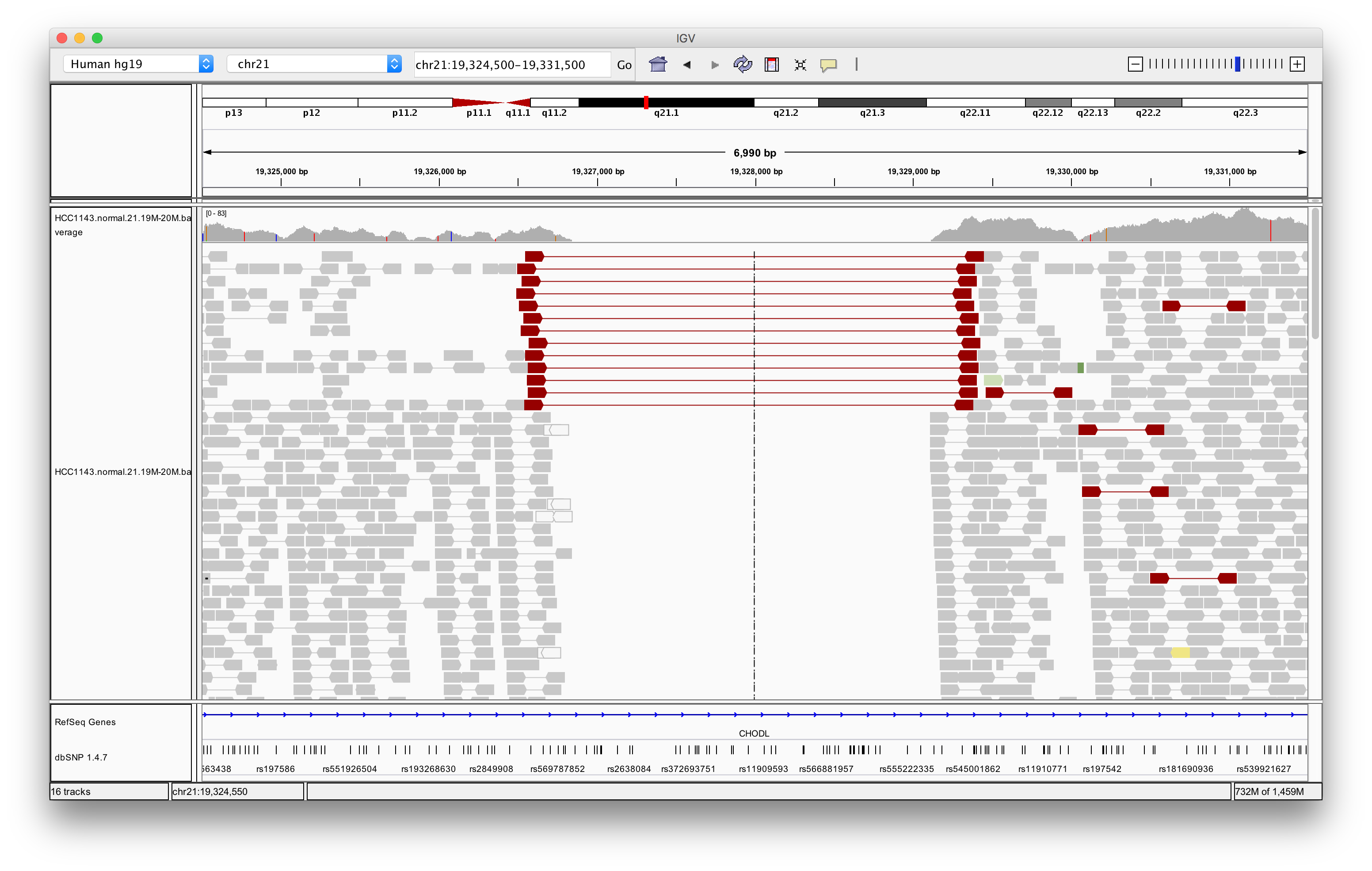
Exercise: What is the insert size of the red flagged read pairs? Can you estimate the size of the deletion?
Answer
The insert size is about 2.8 kb. This includes the reads. The deletion should be about 2.5 - 2.6 kb.