Variant calling
Learning outcomes
After having completed this chapter you will be able to:
- Perform basic calculations regarding the genotype likelihood of individual variants
- Follow
gatkbest practices workflow to perform a variant analysis by:- Calling variants with
gatk HaplotypeCaller - Combining multiple
vcffiles into a singlevcffile
- Calling variants with
- Perform basic operations to get statistics of a
vcffile
Material
The paper on genomic variant call format (gVCF)
GATK best practices germline short variant workflow:
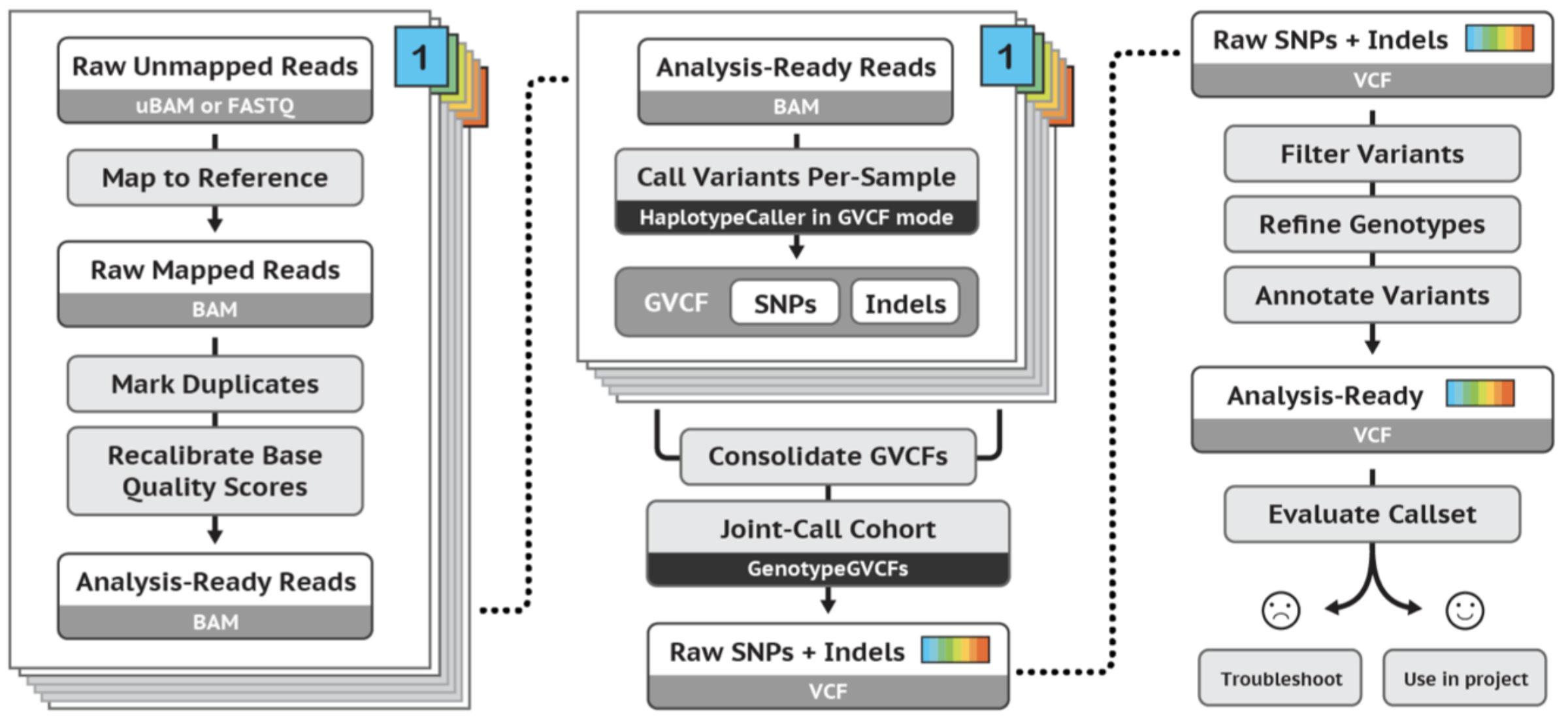
Exercises
1. Variant calling
Calculating PL and GQ by hand
Here’s a function in R to calculate genotype likelihoods as described in Li H. Bioinformatics. 2011;27:2987–93 (assuming equal base error probabilities for all reads):
genotype_likelihood <- function(m,g,e,ref,alt){
(((m-g)*e+g*(1-e))^alt * ((m-g)*(1-e)+g*e)^ref)/(m^(ref+alt))
}
Where:
m: ploidyg: number of alternative allelese: base error probabilityref: number of reference alleles countedalt: number of alternative alleles counted
Exercise: In the scripts directory, create a script called calculate_genotype_likelihoods.R. Copy-paste the above function to the script, and use it to calculate the three genotype likelihoods (for g = 0, g = 1 and g = 2) for a case where we count 22 reference alleles and 4 alternative alleles (so a coverage of 26), and base error probability of 0.01. Calculate the PL values (-10*log10(likelihood)) for each genotype.
Using VScode with R
In order to easily interact with your R script, you can do the following:
- Open the R script in VS code
- In the terminal, type
Rto start the R console - Select the code you’d like to run in the R script
- Type Ctrl+Enter to send it to the console
- After you have finished, type
quit()in the R console.
Answer
# For g = 0 (i.e. 0 alternative alleles)
-10*log10(genotype_likelihood(m = 2, g= 0, e = 0.01, ref = 22, alt = 4))
# [1] 80.96026
-10*log10(genotype_likelihood(m = 2, g= 1, e = 0.01, ref = 22, alt = 4))
# [1] 78.2678
-10*log10(genotype_likelihood(m = 2, g= 2, e = 0.01, ref = 22, alt = 4))
# [1] 440.1746
Exercise: What is the most likely genotype? What is the genotype quality (GQ)? Do you think we should be confident about this genotype call?
Answer
The most likely genotype has the lowest PL, so where g=1 (heterozygous). GQ is calculated by subtracting the lowest PL from the second lowest PL, so 80.96 - 78.27 = 2.69.
This is a low genotype quality (note that we’re in the phred scale), i.e. an error probability of 0.54. This makes sense, if the genotype is heterozygous we would roughly expect to count as many reference as alternative alleles, and our example quite strongly deviates from this expectation.
Calling variants with GATK
The command gatk HaplotypeCaller is the core command of gatk. It performs the actual variant calling.
Exercise: Check out the gatk HaplotypeCaller documentation, and find out which arguments are required.
Answer
Required arguments are:
--input--ouput--reference
Exercise: Generate a script called B10_run_haplotype_caller.sh in B-mother_only. Use it to make a directory called ~/project/results/variants to write the output vcf. In the same script, run gatk HaplotypeCaller with required options on the recalibrated alignment file of the mother (results/bqsr/mother.recal.bam). We’ll focus on a small region, so add --intervals chr20:10018000-10220000.
Answer
#!/usr/bin/env bash
cd ~/project
mkdir -p results/variants
gatk HaplotypeCaller \
--reference data/reference/Homo_sapiens.GRCh38.dna.chromosome.20.fa \
--input results/bqsr/mother.recal.bam \
--output results/variants/mother.HC.vcf \
--intervals chr20:10018000-10220000
Exercise: You can get the number of records in a vcf with piping the output of grep -v '^#' to wc -l. Get the number of variants in the vcf.
Answer
grep -v '^#' variants/mother.HC.vcf | wc -l
Shows you that there are 411 variants in there.
You can get some more statistics with gatk VariantsToTable. The output can be used to easily query things in R or MS Excel.
Here’s an example:
gatk VariantsToTable \
--variant variants/mother.HC.vcf \
--fields CHROM -F POS -F TYPE -GF GT \
--output variants/mother.HC.table
Exercise: Run the command from within a script called B11_variants_to_table.sh, and have a look at the first few records (use e.g. head or less). After that, report the number of SNPs and INDELs.
Answer
Your script should look like:
cd ~/project
gatk VariantsToTable \
--variant results/variants/mother.HC.vcf \
--fields CHROM -F POS -F TYPE -GF GT \
--output results/variants/mother.HC.table
You can get the number of SNPs with:
grep -c "SNP" variants/mother.HC.table
which will give 326
And the number of INDELs with:
grep -c "INDEL" variants/mother.HC.table
that outputs 84
A more fancy way to this would be:
cut -f 3 variants/mother.HC.table | tail -n +2 | sort | uniq -c
Giving:
84 INDEL
1 MIXED
326 SNP
Now, we will perform the variant calling on all three samples. Later we want to combine the variant calls. For efficient merging of vcfs, we will need to output the variants as a GVCF. To do that, we will use the option --emit-ref-confidence GVCF. Also, we’ll visualise the haplotype phasing with IGV in the next section. For that we’ll need a phased bam. You can get this output with the argument --bam-output.
Exercise: Create a script in C-all_samples called C06_run_haplotypecaller.sh. Use it to run gatk HaplotypeCaller for mother, father and son in a loop. Use the same arguments as in the previous exercise. On top of that, add the arguments --emit-ref-confidence GVCF and --bamoutput <phased.bam>.
Answer
#!/usr/bin/env bash
cd ~/project
for SAMPLE in mother father son
do
gatk HaplotypeCaller \
--reference data/reference/Homo_sapiens.GRCh38.dna.chromosome.20.fa \
--input results/bqsr/"$SAMPLE".recal.bam \
--output results/variants/"$SAMPLE".HC.g.vcf \
--bam-output results/variants/"$SAMPLE".phased.bam \
--intervals chr20:10018000-10220000 \
--emit-ref-confidence GVCF
done
2. Combining GVCFs
Now that we have all three GVCFs of the mother, father and son, we can combine them into a database. We do this because it enables us to later add GVCFs (with the option --genomicsdb-update-workspace-path), and to efficiently combine them into a single vcf.
You can generate a GenomicsDB on our three samples like this:
#!/usr/bin/env bash
cd ~/project
gatk GenomicsDBImport \
--variant results/variants/mother.HC.g.vcf \
--variant results/variants/father.HC.g.vcf \
--variant results/variants/son.HC.g.vcf \
--intervals chr20:10018000-10220000 \
--genomicsdb-workspace-path results/genomicsdb
Exercise: Create a script called C07_create_genomicsdb.sh to run this command to generate the database.
You can retrieve the combined vcf from the database with gatk GenotypeGVCFs.
#!/usr/bin/env bash
cd ~/project
gatk GenotypeGVCFs \
--reference data/reference/Homo_sapiens.GRCh38.dna.chromosome.20.fa \
--variant gendb://results/genomicsdb \
--intervals chr20:10018000-10220000 \
--output results/variants/trio.vcf
Exercise: Create a script called C08_genotype_gvcfs.sh to run this command to generate the combined vcf.