Variant Effect Predictor (VEP)
A Practical Introduction
2025-12-12
Somatic vs Germline
- A germline mutation is inherited by the individual from birth. They can be oncogenic (ie Rb = retinoblastoma)
- A somatic mutation, or acquired mutation, happens in somatic cells instead of germ cells and will not pass to offspring (ie TP53).
.png)
How do mutations appear?
- Mutations occur due to replication errors (1 nucleotide per ~10^4) but with reparation mechanisms (1 error per 10^7 - 10^9)
- As a consequence of DNA damage (~70,000 nucleotide lesions or modifications per day)
- Exposure to mutagens such as UV, smoking increases the mutations’ frequency
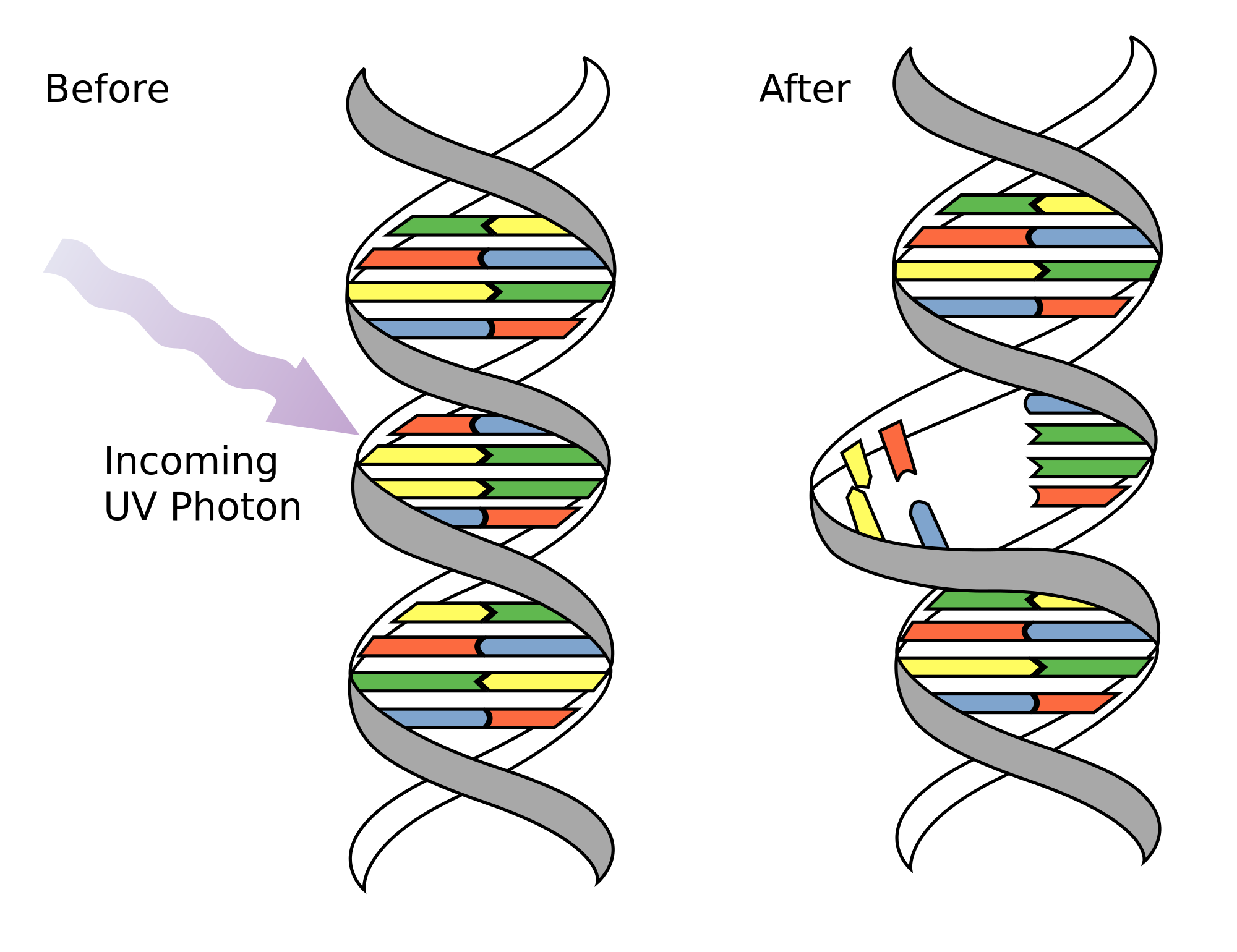
Can the cell repair them?
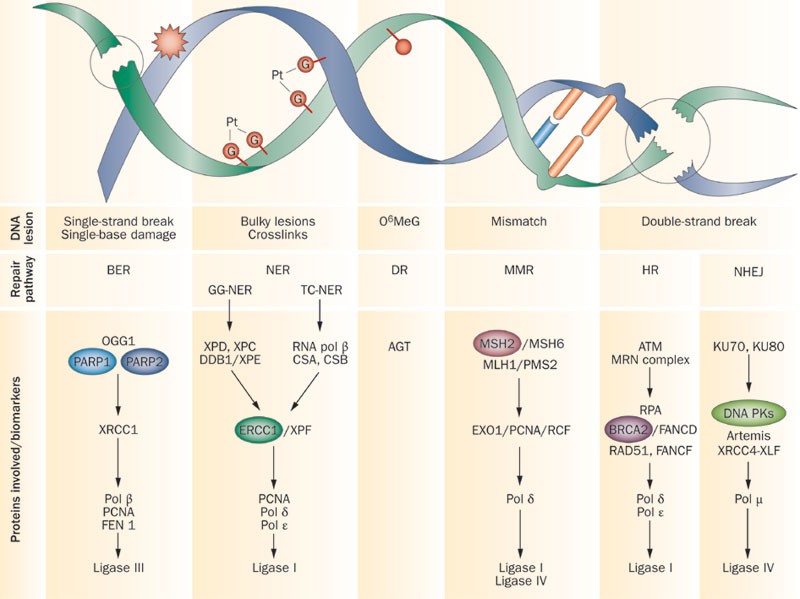
Yes, there are many mechanisms of DNA repair. Some of the most common are:
NER (Nucleotide Excision Repair): Repairs bulky lesions such as thymine dimers caused by UV radiation. This mechanism involves the removal of a short single-stranded DNA segment containing the damage, followed by DNA synthesis using the complementary strand as a template.
MMR (Mismatch Repair): Fixes replication errors such as base mismatches or insertion/deletion loops. MMR recognizes the newly synthesized strand and corrects errors by removing the incorrect nucleotides and replacing them.
BER (Base Excision Repair): Repairs single-base lesions such as oxidative damage, alkylation, or deamination. This involves the removal of damaged bases by specific glycosylases, followed by the excision of the resulting abasic site.
HR (Homologous Recombination): Repairs double-strand breaks using a homologous sequence as a template, typically from a sister chromatid. This is an error-free repair mechanism.
NHEJ (Non-Homologous End Joining): Repairs double-strand breaks without the need for a homologous template. While quicker, this method is error-prone and can lead to insertions or deletions.
Visualizing DNA Repair Mechanisms
Acquisition of mutation in cancer
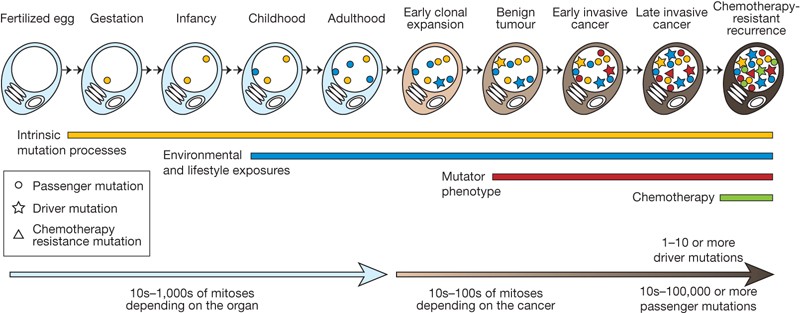
Figure 3: Cancer somatic mutations, The cancer genome, Stratton et al, 2009
VEP (Variant Effect Predictor)
(a) Ensembl-VEP
VEP Impact Categories
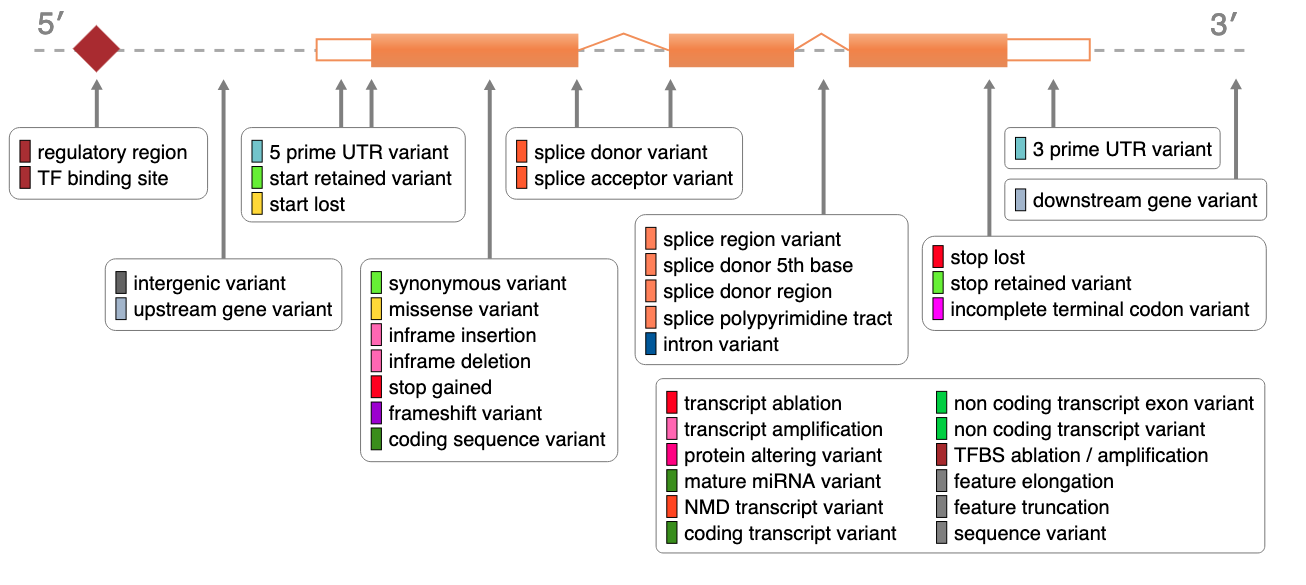
| Impact | Description | Examples |
|---|---|---|
| HIGH | Disruptive. Likely loss of function. | Frameshift, Stop Gained, Splice acceptor/donor. |
| MODERATE | Non-disruptive change to protein. | Missense, In-frame indel. |
| LOW | Unlikely to change function. | Synonymous, Splice region (non-canonical). |
| MODIFIER | Non-coding / Regulatory. | Intronic, UTRs, Intergenic. |
Important Exceptions
Not all HIGH impact = pathogenic
Not all MODERATE impact = benign (e.g., IDH1 p.R132H is LOW but a critical driver mutation)
Always consider biological context!
Exercises

Giphy