Variant Effect Predictor (VEP)
A Practical Introduction
Introduction
Somatic vs Germline
- A germline mutation is inherited by the individual from birth. They can be oncogenic (ie Rb = retinoblastoma)
- A somatic mutation, or acquired mutation, happens in somatic cells instead of germ cells and will not pass to offspring (ie TP53).
.png)
How do mutations appear?
- Mutations occur due to replication errors (1 nucleotide per ~10^4) but with reparation mechanisms (1 error per 10^7 - 10^9)
- As a consequence of DNA damage (~70,000 nucleotide lesions or modifications per day)
- Exposure to mutagens such as UV, smoking increases the mutations’ frequency
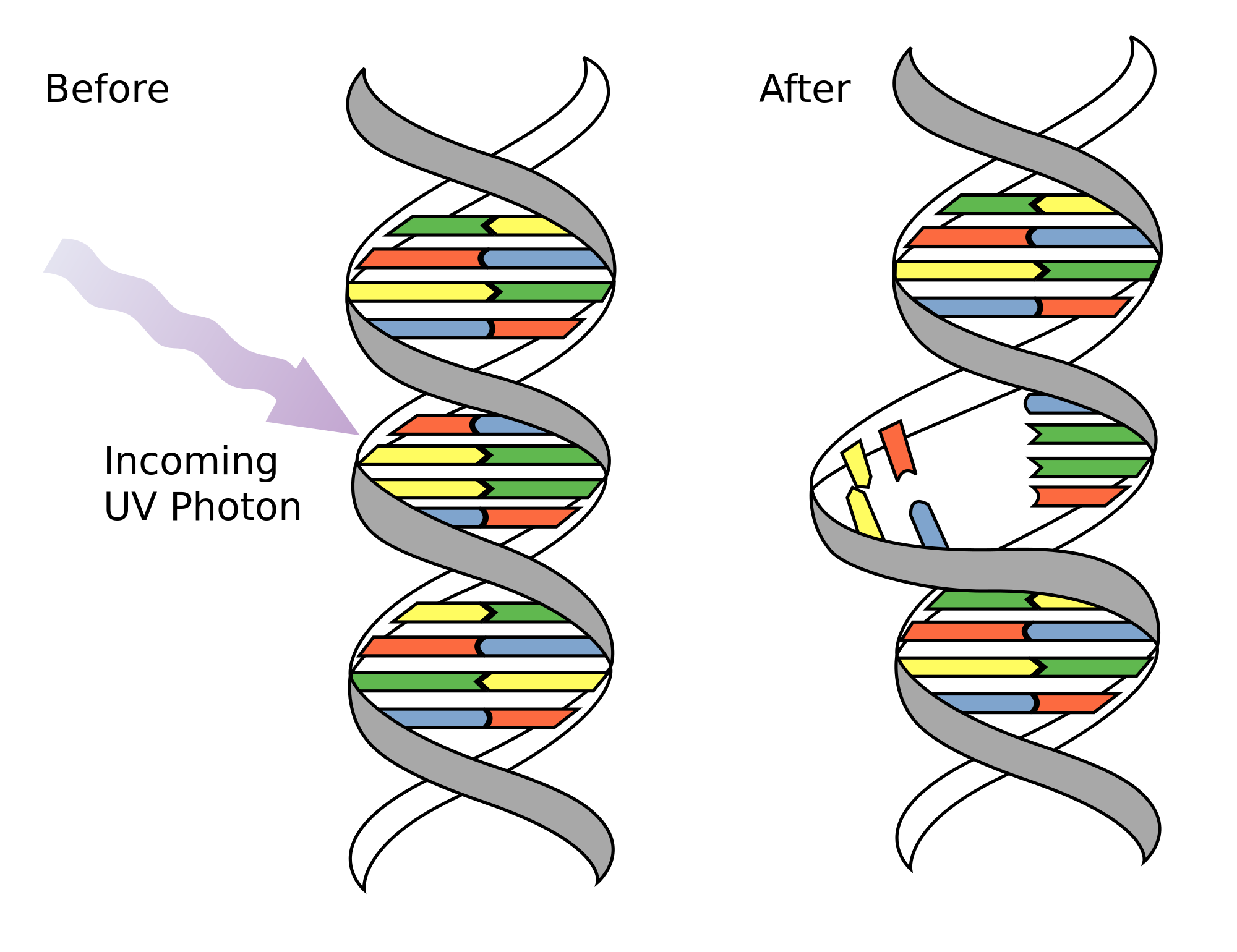
Can the cell repair them?
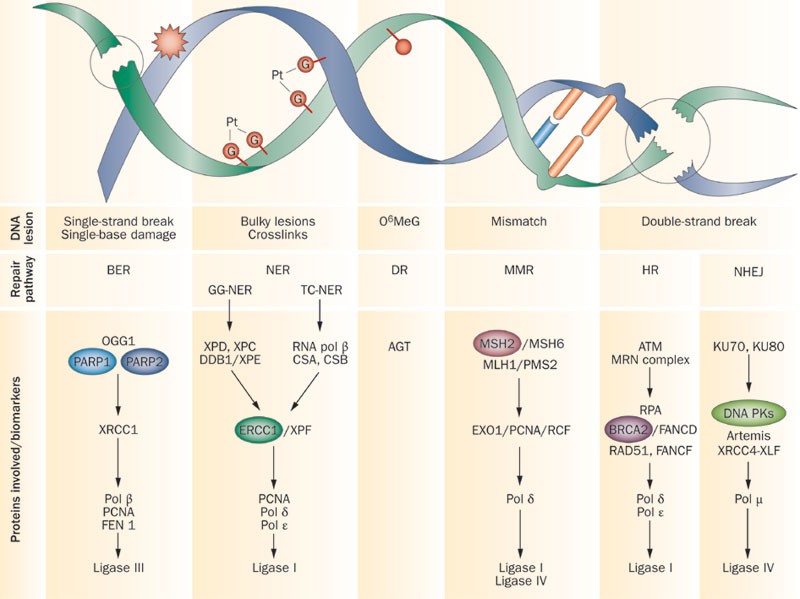
Yes, there are many mechanisms of DNA repair. Some of the most common are:
NER (Nucleotide Excision Repair): Repairs bulky lesions such as thymine dimers caused by UV radiation. This mechanism involves the removal of a short single-stranded DNA segment containing the damage, followed by DNA synthesis using the complementary strand as a template.
MMR (Mismatch Repair): Fixes replication errors such as base mismatches or insertion/deletion loops. MMR recognizes the newly synthesized strand and corrects errors by removing the incorrect nucleotides and replacing them.
BER (Base Excision Repair): Repairs single-base lesions such as oxidative damage, alkylation, or deamination. This involves the removal of damaged bases by specific glycosylases, followed by the excision of the resulting abasic site.
HR (Homologous Recombination): Repairs double-strand breaks using a homologous sequence as a template, typically from a sister chromatid. This is an error-free repair mechanism.
NHEJ (Non-Homologous End Joining): Repairs double-strand breaks without the need for a homologous template. While quicker, this method is error-prone and can lead to insertions or deletions.
Visualizing DNA Repair Mechanisms
Acquisition of mutation in cancer
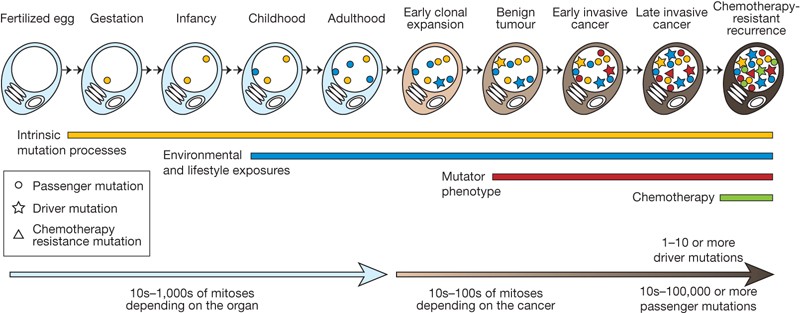
Figure 3: Cancer somatic mut
Concept of driver mutation
Some mutations are more important than others for tumor progression. Perhaps they are more disruptive and detrimental for the cell to harbor. Those can be observed in multiple cancer types (think of TP53 or BRCA1/2).
Not all the driver mutations are known and not all the driver mutations are really always drivers.
What Defines a Driver Mutation?
Not all mutations are driving the tumorigenesis!
Functional Impact:
- Driver mutations confer a selective growth advantage to cells, promoting tumor development and progression. These mutations often affect genes regulating cell cycle, apoptosis, DNA repair, and immune evasion.
- Example pathways: MAPK, PI3K/AKT, and WNT signaling.
Recurrent Patterns Across Tumors:
- Frequently observed across different cancer types (e.g., TP53 in over 50% of cancers).
- Some drivers are specific to tissue types (e.g., EGFR in lung cancer, KIT in gastrointestinal stromal tumors, IDH1 in gliomas).
Types of Driver Mutations
- Oncogenes:
- Gain-of-function mutations in genes like KRAS, BRAF, and EGFR drive tumor growth by promoting uncontrolled cell division or survival.
- Tumor Suppressors:
- Loss-of-function mutations in genes like TP53, RB1, and PTEN impair cellular mechanisms that prevent tumor formation.
- Mutator Genes:
- Mutations in genes like MLH1 or MSH2 lead to genomic instability, enabling the accumulation of additional mutations.
How can we identify what’s a mutation’s role?
- Can we somehow quantify its importance?
VEP (Variant Effect Predictor)
(a) Ensembl-VEP
What is VEP?
- 2010: The first version of VEP was introduced as part of the Ensembl project.
- 2012-2015 Functional prediction scores were introduced.
- 2017 Other databases are now part of the VEP, such as COSMIC, ClinVar, gnomAD. The tool was made more generalizable and personalizable with the addition of VEP’s plugin feature.
- VEP is 100% written in perl VEP Github
Important databases
COSMIC
What it tells us: Somatic mutations found in cancer Example output: COSV59384583; OCCURENCE=1(skin) Interpretation: Mutation seen in skin cancer; frequency helps assess if likely driverClinVar
What it tells us: Clinical significance of variants
Example output: Pathogenic/Likely_pathogenic
Interpretation: Strong evidence for disease causationgnomAD
What it tells us: Population frequency
Example output: AF=0.00001
Interpretation: Very rare variant (<0.01% frequency)AlphaMissense
What it tells us: Predicted functional impact
Example output: likely_pathogenic
Interpretation: AI model predicts damaging effect
What does VEP do?
Imagine thousands of variants to annotate manually to understand what each of them does, searching in different databases, searching in the literature. It would take weeks and it would be error prone. Automation of these tasks reduces the time-to- discovery
- Powerful tool for annotating genomic variants (both somatic and germline)
- Essential in cancer genomics and clinical applications
- Helps understand functional impact of mutations
What is VEP NOT doing for you?
VEP is a great tool, but it does not remove the work from the scientists. Automation simplifies the process but it can give false positives and false negatives. If the experimental setup is low quality, the results most likely will be low quality too.
- Calling variants
- Magic bullet
- Knowing everything
What is not well understood?
There are no databases investigating those aspects of biology that might play an important role in the cancer development. After all it is estimated that >97% of all the mutations are “passenger events” and they do not have direct impact on the tumor growth.
- The effects of epigenetic changes
- Glycosilations
- Transposomes (like LINE-1)
- SV (inversion for example)
- Alternative splicing impacts
- Non-coding RNAs and their alterations
- Tumor Microenvironment
- Microbiome
- Etc.
In summary: Why do we use VEP?
We can answer questions like:
- How damaging is a certain somatic mutation?
- What is the impact in a particular cancer type?
- Is the mutation known for that cancer?
- Are there therapeutic implications?
Installation & Setup
- Installer Script
- Manual Installation
- Docker
- Conda/Bioconda
Installation using conda
# Installing Conda #https://docs.anaconda.com/miniconda/
mkdir -p ~/miniconda3
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda3/miniconda.sh
bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
rm ~/miniconda3/miniconda.sh
source ~/miniconda3/bin/activate
conda init --all
# Using Conda
conda create -n vep samtools python=3.10 ensembl-vep=113
# conda install -c bioconda ensembl-vep # if installing in current env
# Activate VEP's env
conda activate vepVEP’s Basic Configuration
# Test installation
vep --help
# Download cache files, it takes a long time and ~/.vep (~25GB)
vep_install -a cf -s homo_sapiens -y GRCh38 # download precomputed human data
# if install official plugins
vep_install -a p --PLUGINS list # a for action LIST akk plugins
# vep_install -a p --PLUGINS all # install all plugins
# vep_install -a p --PLUGINS dbNSFP,... # install specific plugins
if you want to specify a specific folder to store the VEP’s cache: -c ~/vep_cache
Example plugin installation
SubsetVCF for example does not need additional data, it works directly on the VEP’s output
- installing "SubsetVCF"
- add "--plugin SubsetVCF" to your VEP command to use this plugin
- OKAnd others like REVEL need additional data to work properly
- installing "REVEL"
- This plugin requires data
- See Plugins/REVEL.pm for details
- OKVEP Plugins: Manual install
Plugins can enhance VEP’s capabilities and can add additional depth of information to the annotations (with the price of more complexity).
If this command does not work
Please use this manual option
Core Concepts
Transcript Selection
VEP options for handling multiple transcripts:
--pick: One consequence per variant--pick_allele: One per variant allele--pick_allele_gene: One per variant allele per gene--per_gene: One per gene--flag_pick: Flags selected while keeping others
Consequence Priority
Top 10 most severe consequences:
- Transcript ablation
- Splice acceptor/donor variant
- Stop gained
- Frameshift variant
- Stop lost
- Start lost
- Transcript amplification
- Inframe insertion/deletion
- Missense variant
- Protein altering variant
VEP consequences prediction
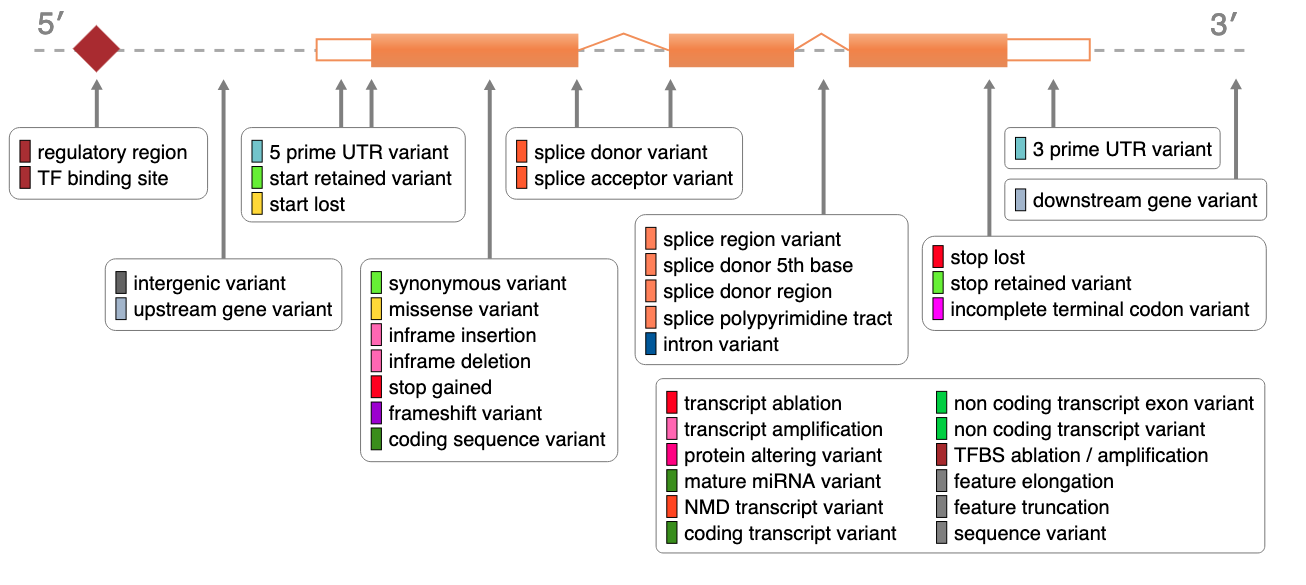
VEP’s consequences
SIF (Sequence Ontology Impact Factors) Categories
HIGH Impact:
Likely to cause a severe effect on protein structure/function.
- Examples:
- Frameshift variants
- Nonsense mutations (e.g. stop codon gained)
- Essential splice-site mutations (e.g. disruption of canonical splice sites).
MODERATE Impact:
Non-disruptive changes that might affect protein structure or function.
- Examples:
- Missense variants (amino acid substitutions).
- In-frame insertions or deletions.
LOW Impact:
Variants that are less likely to have a significant effect on protein function.
- Examples:
- Synonymous changes (no amino acid alteration, [Redundancy]).
- Changes in non-coding regions (outside essential regulatory domains).
MODIFIER Impact:
Usually non-coding or intergenic variants with no expected impact on the protein but might influence gene regulation.
- Examples:
- Intronic variants (non-coding).
- Variants in 5’ or 3’ UTR regions.
Interpretation of HIGH vs. MODERATE Impact
- HIGH: Typically results in loss of protein function or dominant-negative effects. These mutations are often more critical for cancer development and progression (e.g., TP53 inactivation).
- MODERATE: Produces proteins with altered but functional activity, often conferring selective growth advantages under specific contexts (e.g., KRAS or PIK3CA oncogenic activation).
PolyPhen-2 (another predictor included in VEP):
- Predicts impact of amino acid substitutions
- Analyzes both structure and evolutionary conservation
- Specific to human proteins
- Scores range from 0.0 (benign) to 1.0 (damaging)
Categories:
- Probably Damaging (score > 0.85)
- High confidence prediction of affecting protein function
- Possibly Damaging (score 0.15-0.85)
- Moderate prediction of damaging effects
- Benign (score < 0.15)
- Likely to lack phenotypic effect
Practical Exercises
Example 1: Basic Analysis
Input VCF:
Results:
Then we get something more informative like:
- BRAF: p.Val600Glu (melanoma)
- TP53: p.Arg248Trp (multiple cancers)
- BRCA2: missense variant
Download databases’ data
For ClinVar
For COSMIC
We cannot provide those files (a license is required), you need to register on the COSMIC’s website and download the files. For example: CosmicMutantExport.tsv.gz.
You can register here
Example Command for Somatic Analysis
instead of symbol one can select --hgvs or both --hgvs \--symbol
Exercise 2: Cancer Analysis
Adding more specific annotations:
- sift: SIFT is an algorithm for predicting whether a given change in a protein sequence will be deleterious to the function of that protein
- polyphen: PolyPhen-2 predicts the effect of an amino acid substitution on the structure and function of a protein using sequence homology only for human
Advanced Topics
VEP Plugins
Essential plugins for cancer analysis:
REVEL
REVEL paper: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants
You can download here the data (~6.5GB): https://sites.google.com/site/revelgenomics/downloads or https://zenodo.org/records/7072866
Prepare for GRCh38
Usage:
AlphaMissense
AlphaMissense’s Paper: Accurate proteome-wide missense variant effect prediction with AlphaMissense
Prepare for GRCh38
Run it with VEP
dbNSFP
dbNSFP v4 paper: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs
A VEP plugin that retrieves data for missense variants from a tabix-indexed dbNSFP file.
Prepare the data
Prepare for GRCh38
Run it with VEP
Filter Variants with VEP
You can use the tool that is included in the VEP’s suite of tools. This tool generally works very well with data that have been VEP-annotated
Filter SIFT deleterious events `filter_vep -i variant_effect_output.txt -filter "SIFT is deleterious" | grep -v "##" | head -n5
Can be used with pipes, for example (might save memory)
Notes
There are many other plugins that can be used depending on the context and the specific biological question at hand. You can have a look here: Plugins for VEP
You can run multiple plugins at the same time
The more plugins the more computationally expensive it could become
There is a nice
helpincluded in VEP that can be useful for consultation
Best Practices
Quality Control
- Filter low-quality variants before annotation
- Use matched normal samples when available
- Consider sequencing artifacts
- Document filtering criteria
- Use IGV for confirmation
Annotation Strategy
- Use multiple prediction algorithms
- Consider tissue-specific expression
- Include population frequencies
- Add clinical annotations
- Follow standardized guidelines
Points to keep in mind
- Using not only the HIGH impact somatic mutations but also the MODERATE
- Remembering that the “driver” mutation is not necessarily fully representative, we simply do not know many of them
- CNVs (Copy number variants) calling is hard, especially for WES data (WES is only 2% of a genome)
- No tool is perfect! This is especially true for CNV calling
- If samples’ tumor purity is too low, or the quality is too low one should be especially careful with the results
- Experimental design is very important
Resources
Useful Links
Questions?
Feel free to drop a line in the chat or to contact us.
Contact Information
- VEP Support: https://www.ensembl.org/Help/Contact
- Documentation: http://www.ensembl.org/info/docs/tools/vep/
- GitHub: https://github.com/Ensembl/ensembl-vep
- Bioconductor: https://bioconductor.org/