Tutorial on Spark for Bioinformatics
This tutorial gives an introduction to Apache Spark in Scala taking as use case protein sequences and amino acids, commonly used in bioinformatics. The same exercises can also be done with genomic data using nucleotides (A,C,G,T) and the code can be adapted to Python, Java or R.
Download the dataset
The dataset used for this tutorial corresponds to all Swiss-Prot sequences manually reviewed by curators until August 2017: swissprot-aug-2017.tsv.gz (66.0 MB compressed, 200 MB uncompressed, 555’100 sequences or lines).
The data size is good enough to teach the basics of Apache Spark and run on a laptop in standalone mode.
There is also a more challenging dataset, that can be downloaded through this link: uniprot-trembl-aug-2017.tsv.gz (16.7GB compressed, 29.7GB uncompressed, 88’032’926 sequences or lines). This data corresponds to the TrEMBL / automatic sequences that were not reviewed by curators until August 2017.
Once uncompressed, you can see that each line corresponds to a protein sequence and the columns are ordered as follows:
accession geneName specie sequence
P01308 INS HUMAN MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCNNote: To make the examples more readable, we use Swiss-Prot organism mnemonics to represent species but we named it specie (instead of species). For a real analysis of species, you should use the taxon identifiers instead.
Setup Spark environment
This tutorial can be followed, either with the spark-shell (recommended way) or spark-notebooks:
- spark-shell: Download Apache Spark (requires to install Java 8). Start a spark shell with 2GB of memory
$SPARK_HOME/bin/spark-shell --driver-memory 2G.- By default Spark uses all CPUs available in the machine, but can be modified with the option –master local[n] option. Example
$SPARK_HOME/bin/spark-shell --master local[2]will only use 2 cores. In case of using a cluster, the spark-shell can be initialized like this:$SPARK_HOME/bin/spark-shell --executor-memory 4G --master spark://$master_hostname:7077
- By default Spark uses all CPUs available in the machine, but can be modified with the option –master local[n] option. Example
- spark-notebook: Download a spark-notebook version, like for example: spark-notebook 0.7.0 from http://spark-notebook.io/. Extract the archive and start the notebook with the command
./bin/spark-notebook. Access the interface at http://localhost:9001/.
Procedure
Read the dataset and learn about RDDs
Resilient Distributed Dataset (RDD) are a collection of elements partitioned across the nodes of the cluster that can be operated in parallel.
- RDDs automatically recover from node failures
- RDDs can be cached in memory (of the workers)
- RDD can be created either by parallelize or from reading a datasource (fileystem, cassandra, hbase, hdfs, amazon s3…)
//Read data first (you need to uncompress the file in this case)
val data = sc.textFile("swissprot-aug-2017.tsv")
//data: org.apache.spark.rdd.RDD[String] = ...
//Or if working with gz file
val data = sc.textFile("swissprot-aug-2017.tsv.gz")Data can be read from directory using wildcards and can even be compressed. For example this is a valid path: sc.textFile("apache-logs/**/*.gz"). It supports HDFS and other file system, …
Note that this operation goes pretty fast and it will even work if the file is not present. If you run on a cluster the file must be accessed by all workers using the same path. To make sure the file is placed in the correct directory, we can ask to show the first line:
//Shows the first line
data.first If you get an error message that says “Input path does not exist:” then you might need to specify the full path of the file (this happens if you didn’t start the spark-shell where the file was)
Classes and functions
Even though it is possible to do complex tasks in a purely functional manner using anonymous functions and lambdas, it is a good practice to define named functions and classes to better understand the code.
//Defines a class to represent the data in TSV
case class Sequence(accession: String, geneName: String, specie: String, sequence: String)
//Reads a line from the TSV file and returns an object from the Sequence class
def readLine(line: String) = {
val parts = line.split("\t");
Sequence(parts(0), parts(1), parts(2), parts(3))
}Transformations and actions
In Spark there are two types of operations: transformations and actions.
-
A transformation (transforms) creates a new RDD from an existing one. They are lazyly evaluated: They are executed on demand, and therefore are CPU friendly.
-
An action will return a non-RDD type. Actions trigger execution and usually CPU time.
| Transformations | Actions |
|---|---|
| map, flatMap, filter | first, take |
| sample, groupByKey | collect (careful with this one!) |
| distinct | count |
| reduceByKey, sortByKey, … | reduce, save, lookupKey, … |
Transformations are added to the DAG (Directed Acyclic Graph). If you are coming from the RDBMS world, you can think about it as the execution plan, returned with the EXPLAIN keyword.
Let’s see some examples:
//In this case map tells how each line should be mapped / transformed (nothing is actually executed)
val sequences = data.map(readLine) //transformation 1
//Shows first accession or specie (this is an action, but does not require to read the whole file)
sequences.first // action
sequences.first.getClass //action: Notice that this is a class and not an RDD
sequences.first.accession //action
sequences.first.specie //action
//If you have initialized the spark-shell with 2G you may be able to store the result in cache (big difference with Hadoop)
sequences.cache //Asks Spark to persist result in memory if possible
//See how many different species we have
sequences.map(s => s.specie).distinct().count //2 transformations and 1 action
//See the species with the most number of sequences
//3 transformations and 1 action
sequences.map(s => (s.specie, 1)).reduceByKey(_ + _).sortBy(-_._2).take(10).foreach(println)
// This is an action, but all rows must be accessed, because file is not indexed yet.
val humanSequences = sequences.filter(s=> s.specie.equals("HUMAN")) //1 transformation
//Count the number of human sequences
humanSequences.count //1 action
//Sequences containing the 'DANIEL' sequence
val sequencesContainingPattern = sequences.filter(s=> s.sequence.contains("DANIEL")).count
//Use any function (alignment, GC content, ..... )
val result = sequences.map(ANY_FUNCTION_HERE)More complex operations / custom functions
Scala objects can also be created with more complex business logic. Those objects are sent to the workers as long as they are serializable.
object Enzyme extends java.io.Serializable {
//Suppose the enzyme recognizes the portion of the following sequence
val cleavageSite = """[ASD]{3,5}[LI][^P]{2,5}""".r
def matchSite(seq: String): Boolean = cleavageSite.findFirstIn(seq).isDefined
}
//Searches for all sequences that the enzyme will recognize
sequences.filter(s => Enzyme.matchSite(s.sequence)).count//Another example with sorting
object PrositePatterns extends java.io.Serializable {
//Glycosylation prosite pattern
val nglycoPattern = """N[^P][ST][^P]""".r
def countNGlycoSites (seq: String) : Integer = nglycoPattern.findAllIn(seq).length
}
//Returns sequences that have more than 100 N-glycosylation sites predicted (this prediction returns many false positives)
sequences.map(s => (s.accession, s.geneName, PrositePatterns.countNGlycoSites(s.sequence))).filter(r => r._3 > 100).sortBy(-_._3).take(10)Example of Trypsin peptide cuts
The Trypsin is an enzyme used in mass spectometry to identify peptides. It cuts the proteins after an Arginine or a Lysine, but not if it is followed by a Proline. This code shows, how to find the top 10 biggest peptides cut by this enzyme for all given sequences. It also shows how to compute the frequency by peptide length (spectrum)
case class Peptide (accession: String, startPosition: Integer, length: Integer)
object Trypsin extends java.io.Serializable{
//The trypsin cuts just after Arginine (R) or Lysine (K), but not with a Proline (P) after
val pattern = """(?!P)(?<=[RK])""".r
def cut (accession: String, sequence: String) : List[Peptide] = {
val startPositions : List[Int] = List(0) ++ pattern.findAllMatchIn(sequence).map(_.start).toList ++ List(sequence.length)
startPositions.sliding(2).map(l => Peptide(accession, l(0) + 1, l(1)-l(0))).toList
}
}
//Check the top 10 biggest peptides for human sequences
humanSequences.flatMap(s => Trypsin.cut(s.accession, s.sequence)).sortBy(s => -s.length).take(10)
//Draw peptide frequency by peptide length
humanSequences.flatMap(s => Trypsin.cut(s.accession, s.sequence)).map(pep => pep.length).countByValue.toList.sortBy(_._1).foreach(p => println(p._1 + "\t" + p._2 ))External dependencies
Any artifact / jar can be added to the spark context. The spark-shell needs to be initialized with –package, and jars will be sent over the network to the workers
//Need to add --packages at the end (corresponding to Java/Scala artifact. Downloaded by sbt automatically and deployed to worker nodes.
$SPARK_HOME/bin/spark-shell --master spark://$master_hostname:7077 --packages "com.fulcrumgenomics:fgbio_2.11:0.2.0"
import com.fulcrumgenomics.alignment.NeedlemanWunschAligner
val seqToAlign = "MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCD"
humanSequences.map(s=> (s.accession, NeedlemanWunschAligner(1, -1, -3, -1).align(seqToAlign, s.sequence).score)).sortBy(-_._2).take(10).foreach(println)DataFrame and SQL
A dataframe is conceptually equivalent to a table in a relational database or a data frame in R/Python, but with richer optimizations under the hood. DataFrames can be constructed from a wide array of sources such as: structured data files, tables in Hive, external databases, or existing RDDs.
Working with dataframes / SQL can be handy for developers with RDMS experience.
Apache Spark DataFrame / SQL reference.
//The use of data frames may require to import SQL context
val sqlContext = new org.apache.spark.sql.SQLContext(sc)
import sqlContext.implicits._
//Transforms the RDD to a DataFrame
val df = sequences.toDF
df.show
// Print the schema in a tree format (you can imagine nested classes inside the Sequence class)
df.printSchema()
df.createOrReplaceTempView("sequences")
val sqlQuery = """SELECT specie, count(*) as cnt
FROM sequences
GROUP BY specie
ORDER BY cnt DESC
LIMIT 10"""
sqlContext.sql(sqlQuery).show
//OR directly
df.groupBy("specie").agg(count("*") as "cnt").orderBy(desc("cnt")).limit(10).show
//Or a more complex example, that also computes the average of length of the sequence
//This query sorts by the average length, you can see the sequence with less amino acids:
//http://www.uniprot.org/uniprot/P84761
val sqlQuery2 = """SELECT specie, count(*) as cnt, avg(length(sequence)) as avgSeqLength
FROM sequences
GROUP BY specie
ORDER BY avgSeqLength ASC LIMIT 10"""Big data file formats
Avro and Parquet are some of the most used file formats in the Big Data ecosystem. Parquet is a column storage format and is particularly interesting because:
- It eliminates I/O for columns that are not part of query.
- It provides great compression as similar data is grouped together in columnar format.
- It can be saved in partitions
//TSV file: swissprot-aug-2017.tsv (200MB)
//Compressed file: swissprot-aug-2017.tsv.gz (66MB)
//Let's see how much space is the equivalent in Parquet
val data = sc.textFile("swissprot-aug-2017.tsv")
case class Sequence(accession: String, geneName: String, specie: String, sequence: String)
def readLine(line: String) = {
val parts = line.split("\t");
Sequence(parts(0), parts(1), parts(2), parts(3))
}
val sequences = data.map(readLine)
//Save into format Parquet format
//swissprot-aug-2017.parquet folder (109 MB)
sequences.toDF.write.format("parquet").save("swissprot-aug-2017.parquet")Open a new shell and count the number of sequences per species and get the top 10:
val sqlContext = new org.apache.spark.sql.SQLContext(sc)
import sqlContext.implicits._
val sequences = sqlContext.read.parquet("swissprot-aug-2017.parquet")
sequences.printSchema()
//Schema is already defined in the parquet format. No need to define class
sequences.groupBy($"specie").agg(count("*") as "cnt").orderBy($"cnt" desc).limit(10).showAs you can see this query is executed almost instantly, because only the specie column needs to be read.
Spark notebook
The Spark Notebook allows performing reproducible analysis with Scala, Apache Spark and the Big Data ecosystem. They offer the possibilty to visualize directly results in a chart.
val data = sc.textFile("swissprot-aug-2017.tsv")
val sequences = data.map(readLine)
val humanSequences = sequences.filter(s=> s.specie.equals("HUMAN"))
val aaFrequency = humanSequences.flatMap(s=> s.sequence.toList).map(aa => (aa, 1L)).reduceByKey(_ + _).sortBy(-_._2)
//The collect or take action will trigger the rendering of a table that can be toggled to a chart
aaFrequency.collect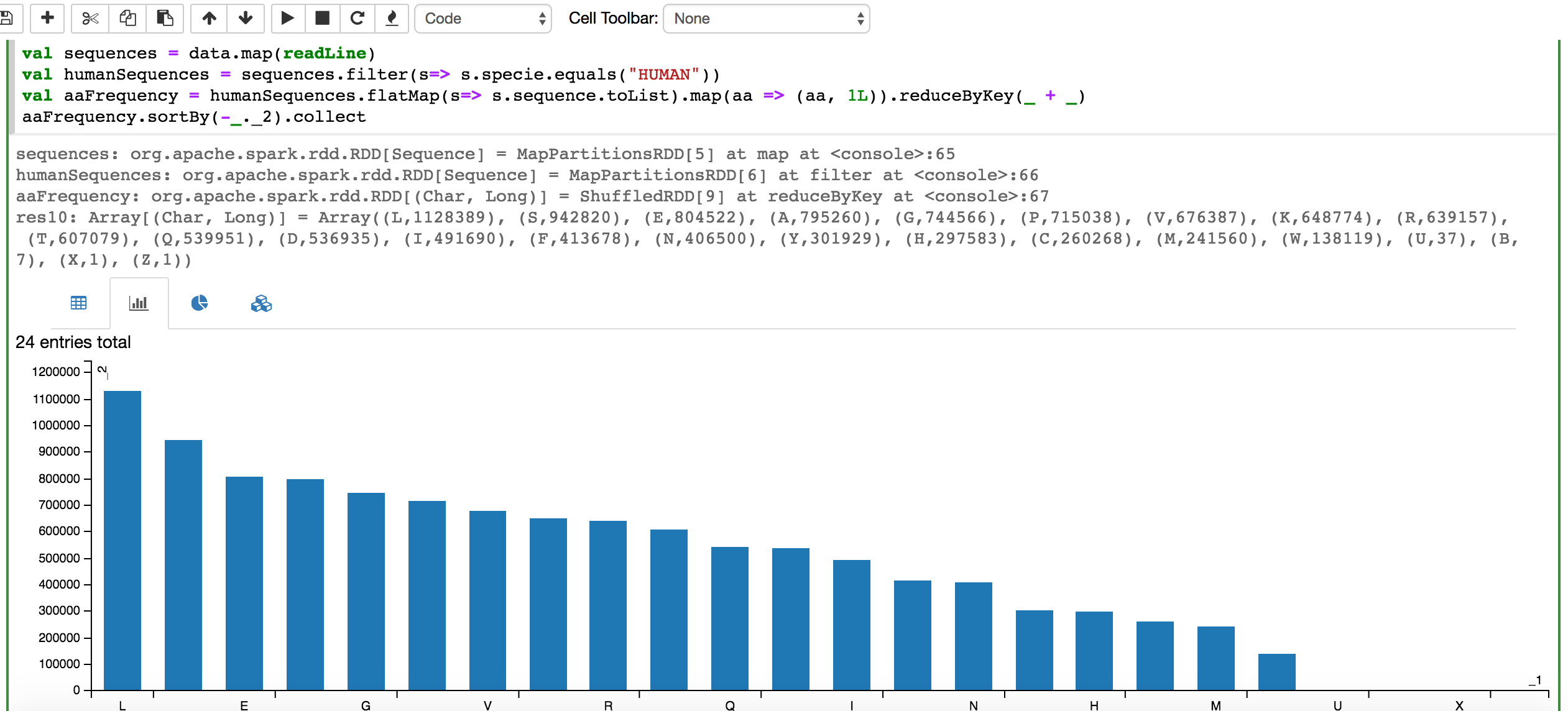
Appendix
ADAM project
ADAM is a genomics analysis platform with specialized file formats built using Apache Avro, Apache Spark and Parquet. Apache 2 licensed.
Look at ADAM project for more complex examples.
FASTA to TSV converter in python
The Python script below was used to convert UniProt FASTA files to TSV format.
import sys
from Bio import SeqIO
fasta_file = sys.argv[1]
for seq_record in SeqIO.parse(fasta_file, "fasta"):
header = str(seq_record.id).split('|')
accessionSpecie = str(header[2]).split("_")
sys.stdout.write(str(header[1])+'\t'+str(accessionSpecie[0])+'\t'+str(accessionSpecie[1])+'\t'+str(seq_record.seq)+ '\n')This post was written by Daniel Teixeira. If you have any suggestions for improvements don’t hesitate to contact him . If you find a typo / bug don’t hesitate to submit a pull request.