Quality control
Learning outcomes
After having completed this chapter you will be able to:
- Find information about a sequence run on the Sequence Read Archive
- Run
fastqcon sequence reads and interpret the results - Trim adapters and low quality bases using
cutadapt
Material
fastqccommand line documentationcutadaptmanual- Unix command line E-utilities documentation
Exercises
Download and evaluate an E. coli dataset
Check out the dataset at SRA.
Exercise: Browse around the SRA entry and answer these questions:
A. Is the dataset paired-end or single end?
B. Which instrument was used for sequencing?
C. What is the read length?
D. How many reads do we have?
Answers
A. paired-end
B. Illumina MiSeq
C. 2 x 251 bp
D. 400596
Now we will use some bioinformatics tools to do download reads and perform quality control. The tools are pre-installed in a conda environment called ngs-tools. Every time you open a new terminal, you will have to load the environment:
conda activate ngs-tools
Make a directory reads in ~/workdir and download the reads from the SRA database using prefetch and fastq-dump from SRA-Tools into the reads directory. Use the code snippet below to create a scripts called 01_download_reads.sh. Store it in ~/workdir/scripts/, and run it.
#!/usr/bin/env bash
cd ~/workdir
mkdir reads
cd reads
prefetch SRR519926
fastq-dump --split-files SRR519926
Exercise: Check whether the download was successful by counting the number of reads in the fastq files and compare it to the SRA entry.
Tip
A read in a fastq file consists of four lines (more on that at file types). Use Google to figure out how to count the number of reads in a fastq file.
Answer
e.g. from this thread on Biostars:
## forward read
echo $(cat SRR519926_1.fastq | wc -l)/4 | bc
## reverse read
echo $(cat SRR519926_2.fastq | wc -l)/4 | bc
Run fastqc
Exercise: Create a script to run fastqc and call it 02_run_fastqc.sh. After that, run it.
Tip
fastqc accepts multiple files as input, so you can use a wildcard to run fastqc on all the files in one line of code. Use it like this: *.fastq.
Answer
Your script ~/workdir/scripts/02_run_fastqc.sh should look like:
#!/usr/bin/env bash
cd ~/workdir/reads
fastqc *.fastq
Exercise: Download the html files to your local computer, and view the results. How is the quality? Where are the problems?
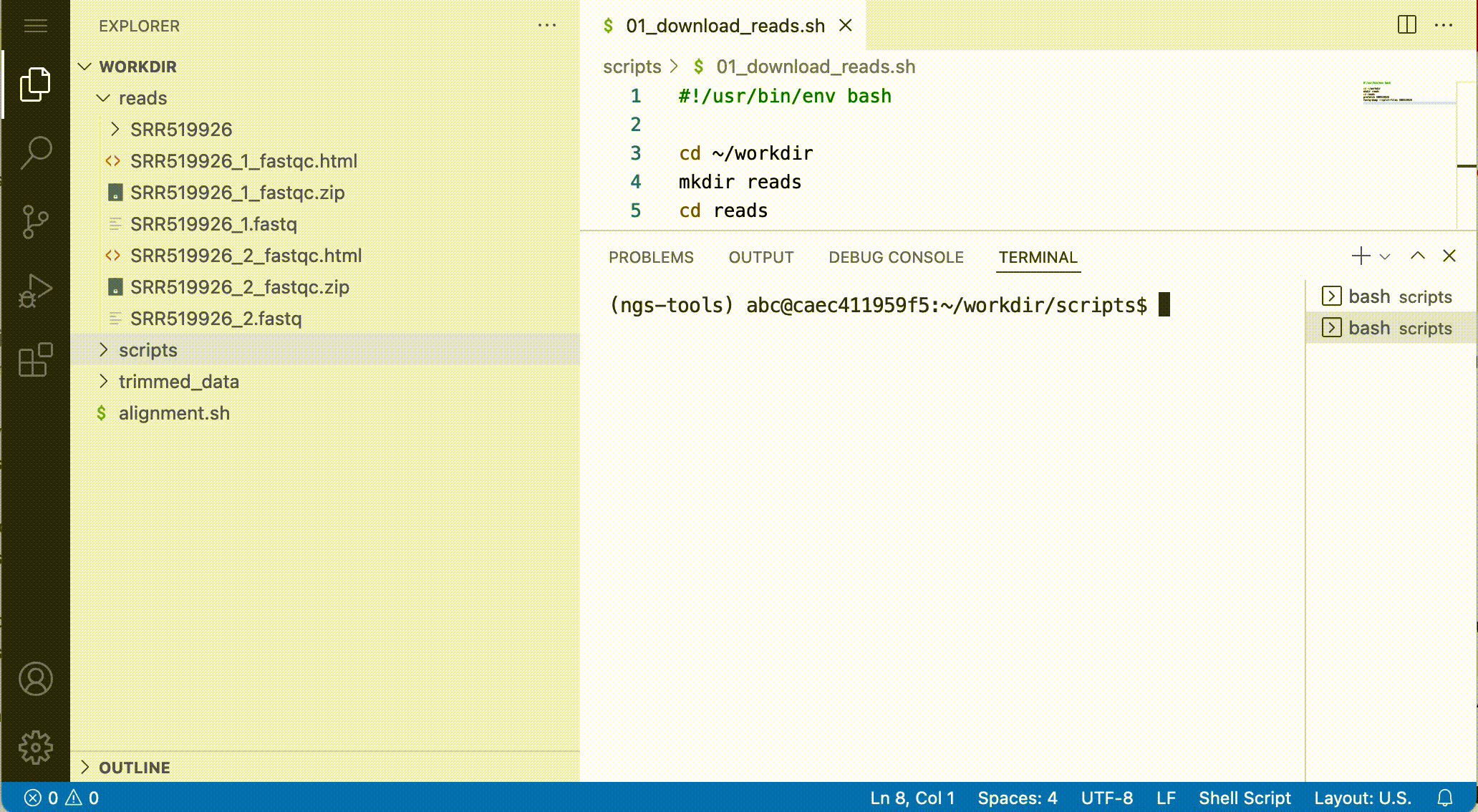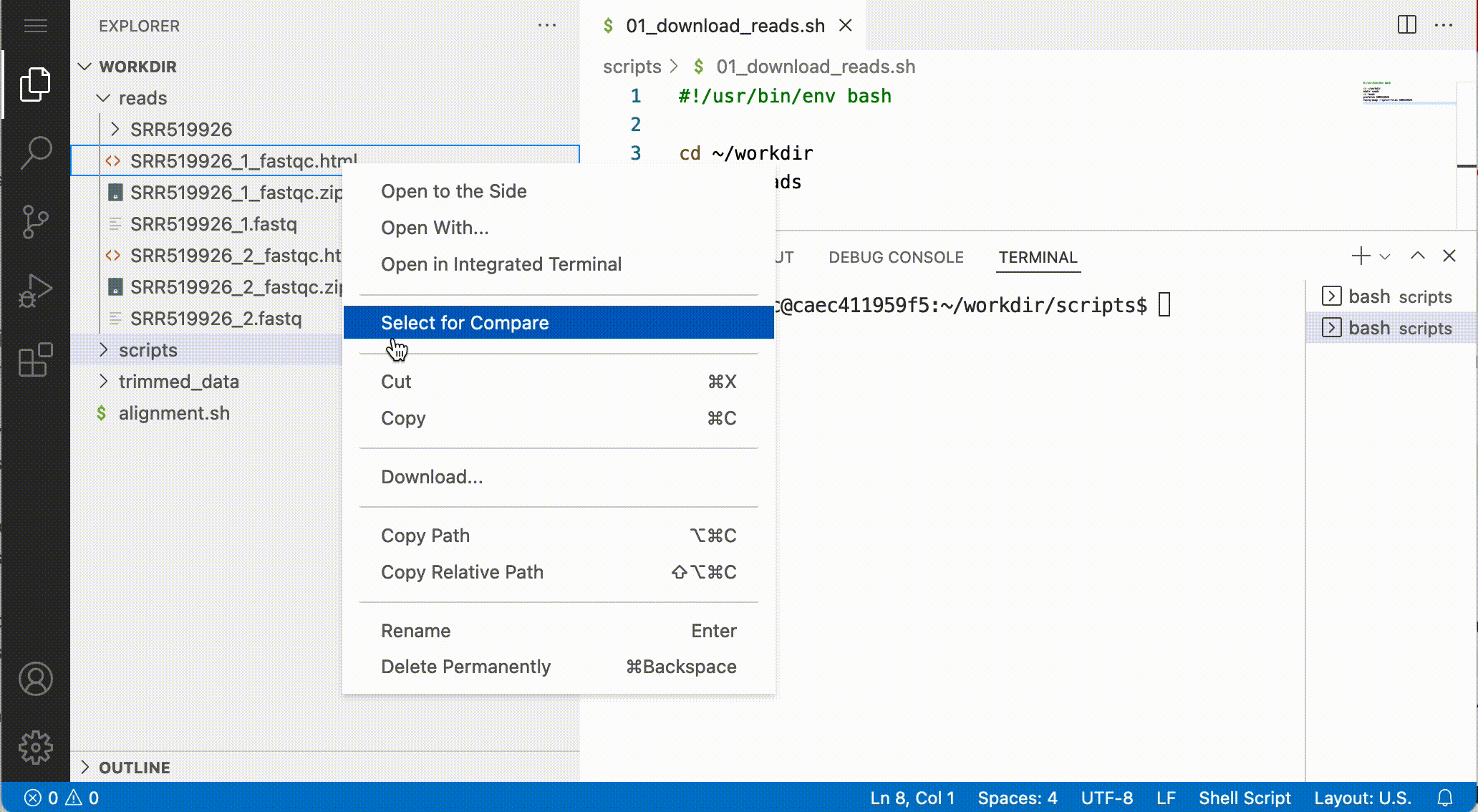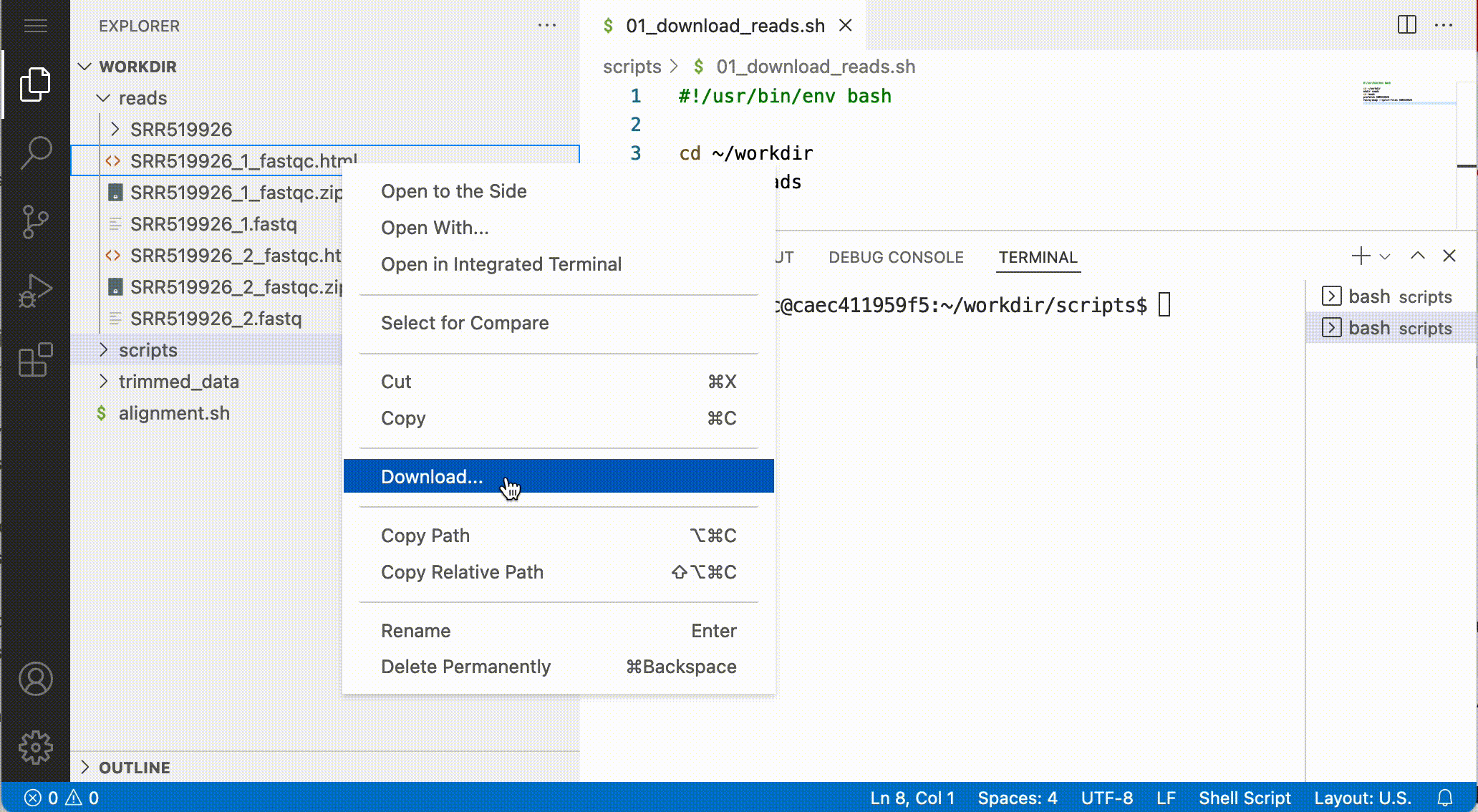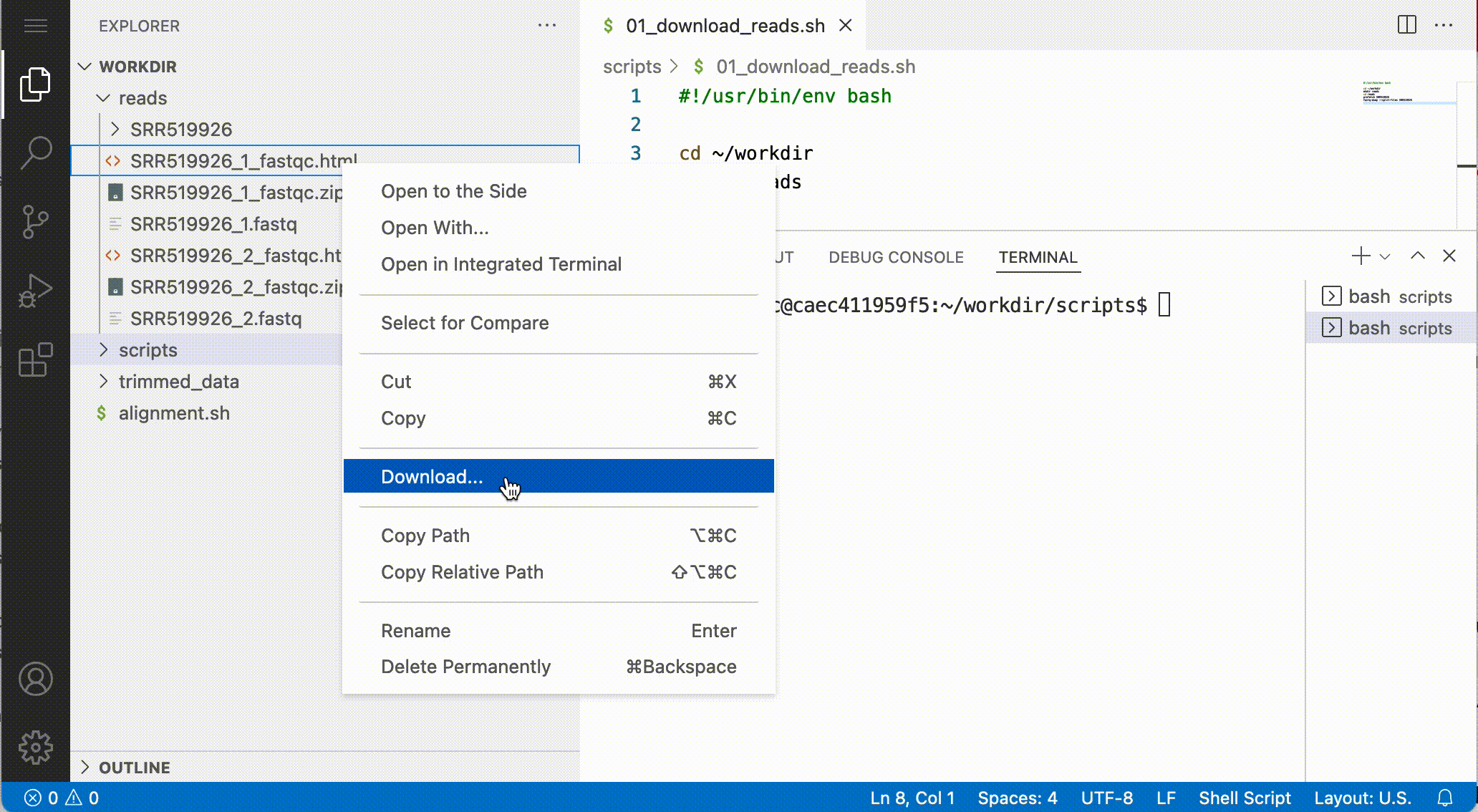
Downloading files
You can download files by right-click the file and after that select Download:
Answer
There seems to be:
- Low quality towards the 3’ end (per base sequence quality)
- Full sequence reads with low quality (per sequence quality scores)
- Adapters in the sequences (adapter content)
We can probably fix most of these issues by trimming.
Trim the reads
We will use cutadapt for trimming adapters and low quality bases from our reads. The most used adapters for Illumina are TruSeq adapters. To run cutadapt you need to specify the adapter sequences with options -a (or --adapter) and -A. A reference for the adapter sequences can be found here.
Exercise: The script below will trim the sequence reads. However, some parts are missing. We want to:
- trim bases with a quality lower then 10 from the 3’ and 5’ end of the reads,
- keep only reads with a read length not shorter than 25 base pairs.
Copy the code below to a script in your scripts directory (~/workdir/scripts) and call it 03_trim_reads.sh. Fill in the missing options that in between []. After that execute the script to trim the data.
Hint
Check out the helper of cutadapt with:
cutadapt --help
#!/usr/bin/env bash
TRIMMED_DIR=~/workdir/trimmed_data
READS_DIR=~/workdir/reads
mkdir -p $TRIMMED_DIR
cutadapt \
--adapter AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \
-A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT \
[QUALITY CUTOFF OPTION] \
[MINIMUM LENGTH OPTION] \
--output $TRIMMED_DIR/paired_trimmed_SRR519926_1.fastq \
--paired-output $TRIMMED_DIR/paired_trimmed_SRR519926_2.fastq \
$READS_DIR/SRR519926_1.fastq \
$READS_DIR/SRR519926_2.fastq
Answer
Your script (~/workdir/scripts/03_trim_reads.sh) should look like this:
#!/usr/bin/env bash
TRIMMED_DIR=~/workdir/trimmed_data
READS_DIR=~/workdir/reads
mkdir -p $TRIMMED_DIR
cutadapt \
--adapter AGATCGGAAGAGCACACGTCTGAACTCCAGTCA \
-A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT \
--quality-cutoff 10,10 \
--minimum-length 25 \
--output $TRIMMED_DIR/paired_trimmed_SRR519926_1.fastq \
--paired-output $TRIMMED_DIR/paired_trimmed_SRR519926_2.fastq \
$READS_DIR/SRR519926_1.fastq \
$READS_DIR/SRR519926_2.fastq
The use of \
In the script above you see that we’re using \ at the end of many lines. We use it to tell bash to ignore the newlines. If we would not do it, the cutadapt command would become a very long line, and the script would become very difficult to read. It is in general good practice to put every option of a long command on a newline in your script and use \ to ignore the newlines when executing.
Exercise: Create a script to run fastqc on the trimmed fastq files called 04_run_fastqc_trimmed.sh, and answer these questions:
A. Has the quality improved?
B. How many reads do we have left?
Answers
Your script 04_run_fastqc_trimmed.sh should look like:
#!/usr/bin/env bash
cd ~/workdir/trimmed_data
fastqc paired_trimmed*.fastq
A. Yes, low quality 3’ end, per sequence quality and adapter sequences have improved.
B. 315904