Being reproducible with Snakemake
Learning outcomes
After having completed this chapter you will be able to:
- Use an input function to work with an unknown number of files
- Run scripts from other languages (Python and R)
- Deploy a rule-specific conda environment
- Deploy a rule-specific Docker/Apptainer container
Material
Workflow from previous session
If you didn’t finish the previous part or didn’t do the optional exercises, you can restart from fully commented snakefiles, with a supplementary .fastq files quality check rule and multithreading, memory usage control implemented in all rules. You can download all the files (workflow and config) here or copy them after clonining the course repository locally:
git clone https://github.com/sib-swiss/containers-snakemake-training.git # Clone repository
cd containers-snakemake-training/ # Go in repository
cp -r docs/solutions_day2/session3 destination_path # Copy folder where needed; adapt destination_path to required path
Exercises
In this series of exercises, you will create the last two rules of the workflow. Each rule will execute a script (one in Python and one in R; don’t worry, this is not a programming course, so we wrote the scripts for you!), and both rules will have dedicated environments that you will need to take into account in the snakefiles.
Development and back-up
During this session as well, you will modify your Snakefile quite heavily, so it may be a good idea to make back-ups from time to time (with cp or a simple copy/paste) or use a versioning system. As a general rule, if you have a doubt on the code you are developing, do not hesitate to make a back-up beforehand.
Creating a rule to gather read counts
To perform a Differential Expression Analysis (DEA), it is easier to have a single file gathering all the read counts from the different samples. The next rule that you will create will both find the required files and merge them, thanks to an input function and a Python script.
Building the general rule structure
We already wrote the common elements of the rule so that you can focus on the most interesting parts (the missing input and the missing elements at the end):
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Here, there is no wildcards in the rule: only one file will be created, and its name will not change depending on sample names because we use all the samples to create it.
Explicit is better than implicit
Even if a software cannot multithread, it is useful to add threads: 1 to keep the syntax consistent between rules and clearly state that the software works with a single thread.
Exercise: Given that this rule and the next one will be quite different from the previous ones, it is a good idea to implement them in a new snakefile. Create a new file workflow/rules/analysis.smk and copy the previous rule structure in it. Do you need to change anything else to use this rule in your workflow?
Answer
To actually use this rule, Snakemake needs to be aware that it exists: this is done with the include statement. We need to add the following lines to the main Snakefile (workflow/Snakefile):
include: 'rules/analyses.smk'
The Snakefile will look like this:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Let’s start filling those missing elements!
Gathering the input files
This task is quite complex: we need a way to identify all the rule inputs and gather them in a Python list. Here, there are only six samples, so in theory, you could list them directly… However, it isn’t good practice and it quickly becomes un-manageable when the number of sample increases. Fortunately, there is a much more elegant and convenient solution: an input function, which provides the added benefit of scaling up very well.
We wrote one for you:
1 2 3 4 5 6 7 | |
Snakemake wildcards vs Python f-strings
This input function is pure Python code: in the return statement, {sample} isn’t a wildcard, it is an f-string variable! This shows that you can natively use basic Python elements in a workflow: Snakemake will still be able understand them. This is because Snakemake was built on top of Python.
This function will loop over the list of samples in the config file and replace {sample} with the current sample name of the iteration to create a string which is the output path from the rule reads_quantification_genes of said sample. Then, it will aggregate all the paths in a list and return this list.
More on input functions
- Input functions take the
wildcardsglobal object as single argument - You can access wildcard values inside an input function with the syntax
{wildcards.wildcards_name} - Input functions can return lists of files, which will be automatically handled like multiple inputs by Snakemake
- Input functions can also return a dictionary; in this case, the function should be called with the
unpack()function:The dictionary keys will be used as input names and the dictionary values will be used as input values, providing a list of named inputsinput: unpack(<function_name>)
- Input functions can also return a dictionary; in this case, the function should be called with the
Input functions and output directory
Input functions are evaluated before the workflow is executed, so they cannot be used to list the content of an output directory, since it does not exist before the workflow is executed. Instead, you can use a checkpoint to trigger a re-evaluation of the DAG.
Exercise: Insert the function get_gene_counts() in workflow/rules/analysis.smk and adapt the input value of count_table accordingly. Do you need to insert the function in a specific location?
Answer
The first step is to add the input function to the file. However, it needs to appear before the rule count_table, otherwise we will see the error name 'get_gene_counts' is not defined. In other words, the function needs to be defined before Snakemake looks for it when it parses the input. Then, we need to set the function name as the rule input value.
The modular snakefile workflow/rules/analysis.smk will resemble this:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 | |
input directive. Doing so would actually change Snakemake behaviour!
Now that the rule inputs are defined, we need to set-up the script to process them.
Using a Python script in Snakemake
Getting the script
The counts will be concatenated thanks to a script called count_table.py. It was written in Python, takes a list of files as input, and produces one output, a tab-separated table containing read counts of the different samples for each gene. You can download it with:
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/docs/solutions_day2/session4/workflow/scripts/count_table.py
Or you can copy it from here:
Click here to see a nice Python script!
| count_table.py | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 | |
Exercise: Get the script with your favourite method and place it the proper folder according to the official documentation. Where should you store it?
Answer
Scripts should be gathered in their own dedicated folder: workflow/scripts.
Deciding how to run the script
If you remember the presentation, there are two directives that you can use to run external scripts in Snakemake: run and script. While both allow to run Python code, they are not equivalent, so there is a choice to make!
Exercise: Check out the script content. Depending on what you find, choose a directive and implement it in place of the last two missing elements (?) of rule count_table.
Script path is relative…
… to the Snakefile calling it. If you followed the recommended workflow structure, modular snakefiles are placed in a rules subfolder (like rules/analysis.smk) and scripts are placed in a scripts subfolder (like scripts/count_table.py). You need to find a path between those two subfolders.
Answer
There are two things to check before deciding which directive to use:
-
The script length:
If we open the script in a text editor or run
wc -l workflow/scripts/count_table.py, we see that it is 67 lines long. It is also quite complex, with function definitions, loops… This favours thescriptdirective, as it’s better to userunwith short and simple code. -
The use of external packages (packages that are not included in a default Python installation):
Another way to put this is: does the script need a special environment to work? If so, then we have to use the
scriptdirective, as it is the only one to accommodate for conda environments or containers. This means that this criteria takes precedence over the previous one: if we need to run a short script within a dedicated environment,scriptis the only way to do it.Here, there are several
importstatements at the top of the script, includingimport pandas as pd # Non built-in package.pandasis a great package, but it is not part of the built-in packages shipped with Python. This means that the script needs a dedicated environment to run and confirm that we need thescriptdirective.
With this in mind, the rule will be:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Using the same Python script in and out of Snakemake
To avoid code redundancy, it would be ideal to have a script that can be called by Snakemake but also work with standard Python (and be used outside Snakemake). The two main ways to do this are:
- Implement the script as a module/package and use this module in Snakemake, for example with a command-line interface in
shell - Test whether the
snakemakeobject exists in the script:- If so, the script can process the Snakemake values
- When the script is launched by Snakemake, there is an object called
snakemakethat provides access to the same objects that are available in therunandshelldirectives (input,output,params,wildcards,log,threads,resources, config). For instance, you can usesnakemake.input[0]to access the first input file of a rule, orsnakemake.input.input_nameto access a named input
- When the script is launched by Snakemake, there is an object called
- If not, the script can use other parameters, for example those coming from command-line parsing
- If so, the script can process the Snakemake values
Providing a rule-specific conda environment
Given the presence of a non-default package in the script, we need to find a solution to make it accessible inside the rule. The easiest way to do that is to create a rule-specific conda environment. In Snakemake, you can do this by providing an environment config file (in YAML format) to the rule with the conda directive.
(Optional) Exercise: If you have time, you can create your own config file for the environment using the tip on ‘Environment features’ below. If you need a reminder on how an environment file look, you can check out slide 19 of the presentation (available here). Otherwise, you can directly skip to the answer.
Environment features
- Environment
nameispy3.12 - It uses two
channels:conda-forgeandbioconda(in that order) - It requires
pythonwith at least version3.12 - It requires
pandaswith version2.2.3exactly - Like with scripts, config files should be stored in their own dedicated folder:
workflow/envs
Answer
The config file, created in workflow/envs/py.yaml should look like this:
1 2 3 4 5 6 7 8 | |
Exercise: Add the conda environment to the rule.
Environment file path is relative…
… to the Snakefile calling it. If you followed the recommended workflow structure, modular snakefiles are placed in a rules subfolder (like rules/analysis.smk) and environment files are placed in a envs subfolder (like envs/py.yaml). You need to find a path between those two subfolders.
Answer
We need to fill the last two missing elements with the directive name, conda, and its value, the script location:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Using conda environments improves reproducibility for many reasons, including version control and the fact that users do not need to manually manage software dependencies. The first workflow execution after adding Conda environments will take more time than usual because snakemake (through conda) has to download and install all the software in the working directory.
Adapting the Snakefile and running the rule
All that is left is running the rule to create the table.
Exercise: Find the snakemake command you should run to create the desired output (which one is it?) and execute the workflow. Is there anything else to do beforehand?
Answer
We cannot launch the workflow directly: first, we need to update rule all input to use the output of rule count_table. After this, your Snakefile should be:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
Finally, run the workflow with:
snakemake -c 4 -p --sdm conda
--sdm conda, otherwise Snakemake will not use the environment you provided.
You should see the rule count_table executed with 6 files as input. You can also check the log of rule count_table to see if the script worked as intended.
Creating a rule to detect Differentially Expressed Genes (DEG)
The final rule that you will create in this course will use an R script to process the global read count table previously created and detect differentially expressed genes. As such, you will see several common elements between this rule and rule count_table (external scripts, dedicated environments, rule structure…) and the process to implement this rule will also be very similar. However, it will be easier as you won’t need to use an input function.
Building the general rule structure (again)
We also wrote the common elements of the rule so that you can focus on the most interesting parts (the missing elements at the end):
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 | |
Once again, we do not need to use wildcards in this rule, because all the files are precisely defined.
Exercise: Given the rule structure above, update the Snakefile so that it creates the final output of the workflow.
Answer
There is only one thing to update in the target rule input:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 | |
differential_expression as inputs of rule all, only one suffices.
As mentioned above, we don’t need an input function because the input of rule differential_expression is easy to identify, so we’ll directly focus on a way to run the R script.
Using an R script in Snakemake
Downloading the script
The DE analyses will be performed thanks to a script called DESeq2.R. It was written in R, takes a read count table as input, and produces two outputs, a tab-separated table containing DEG (and statistical results) and a .pdf file containing control plots of the analysis. You can download it here or with the command:
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/docs/solutions_day2/session4/workflow/scripts/DESeq2.R
Exercise: Download the script and place it the right folder.
Answer
You can place this script in the same folder than the Python script, workflow/scripts. There is nothing in the official documentation about placing scripts from different languages in separate folders, but if you use a large number of scripts, it might be worth considering. You could also gather scripts by topic, similarly to .smk files.
Running the script
The next exercise won’t be as guided as the other ones. This is done on purpose as you have seen everything you need to solve it!
Exercise: Find a way to run the R script and fill the missing elements in the rule.
What do you need to take into account?
- The directive you need to run the script
- The location/path of the script
- Check whether the script need a special environment
- If so, remember a certain Docker image you created yesterday
Answer
Like with the Python script, there are two problems to solve to run the R script:
-
Which directive to use?
There isn’t much of a choice here… If you remember the presentation, there is only one way to run non-Python code in Snakemake: the
scriptdirective:1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
rule differential_expression: ''' This rule detects DEGs and plots control graphs (PCA, heatmaps...). ''' input: table = rules.count_table.output.table output: deg = 'results/deg_list.tsv', pdf = 'results/deg_plots.pdf' log: 'logs/differential_expression.log' benchmark: 'benchmarks/differential_expression.txt' resources: mem_gb = 1 threads: 2 ?: ? script: # Add script directive '../scripts/DESeq2.R' # Add script location relative to rule file -
Does it use external packages and need a specific environment?
If you look at the top of the script, you will see several (11 to be exact!)
library()calls. Each of them imports an external package. All of these could be gathered in a conda environment, however when numerous libraries are involved, it is sometimes easier to use a container. During Day 1 - Session 3 (Working with Dockerfiles), you built your own Docker image, calleddeseq2. This image actually contains everything required by the script:1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
rule differential_expression: ''' This rule detects DEGs and plots control graphs (PCA, heatmaps...). ''' input: table = rules.count_table.output.table output: deg = 'results/deg_list.tsv', pdf = 'results/deg_plots.pdf' log: 'logs/differential_expression.log' benchmark: 'benchmarks/differential_expression.txt' resources: mem_gb = 1 threads: 2 container: # Add container directive 'docker://geertvangeest/deseq2:v1' # Try with your own image; if it doesn't work, use Geert's script: '../scripts/DESeq2.R'
Using the same R script in and out of Snakemake
Inside the script, an S4 object named snakemake, analogous to the Python one, is available and allows access to Snakemake objects: input, output, params, wildcards, log, threads, resources, and config. Here, the syntax follows that of S4 classes with attributes that are R lists. For example, you can access the first input file with snakemake@input[[1]] (remember that in R, indexing starts at 1). Named objects can be accessed the same way, by providing the name instead of an index: snakemake@input[["myfile"]] to access the input called myfile.
Now, all that is left is running the workflow, check its outputs and visualise its DAG!
Running the workflow
Exercise: Run the workflow. How many DEGs are detected during the analysis?
Answer
You can run the workflow with:
snakemake -c 4 -p --sdm apptainer
--sdm apptainer, otherwise Snakemake will not pull the image and the script will be executed in the default environment (which will most likely lead to a crash).
During the run, you should see log messages about Snakemake managing the Docker image:
Pulling singularity image docker://geertvangeest/deseq2:v1.
[...]
Activating singularity image /path/to/snakemake_rnaseq/.snakemake/singularity/8bfdbe93244feb95887ab5d33a705017.simg
To find how many genes are differentially expressed, check out the last output file, results/deg_list.tsv. 10 genes are differentially expressed in total: 4 up-regulated and 6 down-regulated.
Containerisation of Conda-based workflows
Snakemake can also automatically generate a Dockerfile that contains all required environments in a human readable way. If you want to see how a Snakemake-generated Dockerfile looks like, use:
snakemake -c 1 --containerize > Dockerfile
(Optional) Exercise: If you had to re-run the entire workflow from scratch, what command would you use?
Answer
You can re-run the whole workflow with:
snakemake -c 4 -p -F --sdm conda apptainer
-Fforces the execution of the entire workflow- Remember that you need both Conda and Docker-based environments for this run! You can combine
--sdm condaand--sdm apptainerinto a single command--sdm conda apptainer. Otherwise, you will lack some software and packages and the workflow will crash!
Exercise: Visualise the DAG of the entire workflow.
Answer
You can get the DAG with:
snakemake -c 1 -p -F --dag | dot -T png > images/total_dag.png
You should get the following DAG (open the picture in a new tab to zoom in):
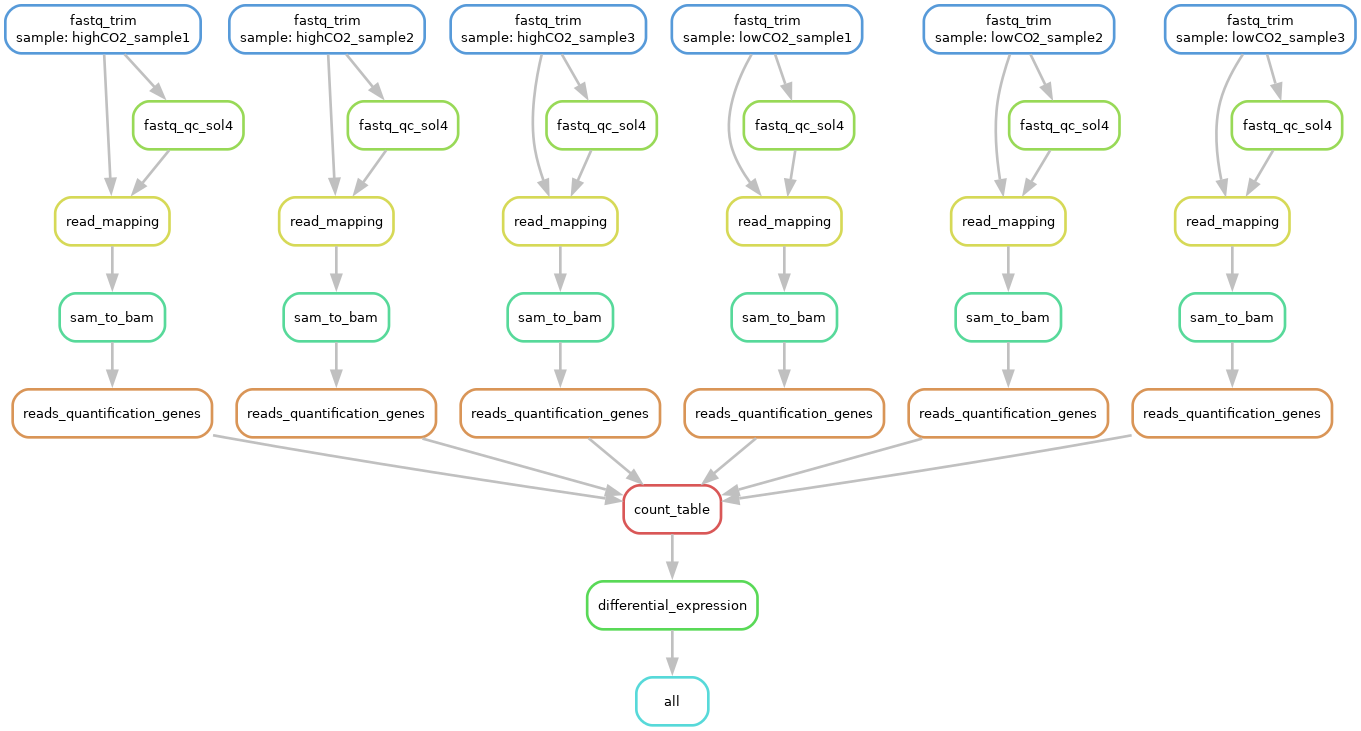
Congratulations, you made it to the end! You are now able to create a Snakemake workflow and make it reproducible thanks to Conda and Docker/Apptainer! To make things even better, have a look at Snakemake’s best practices!