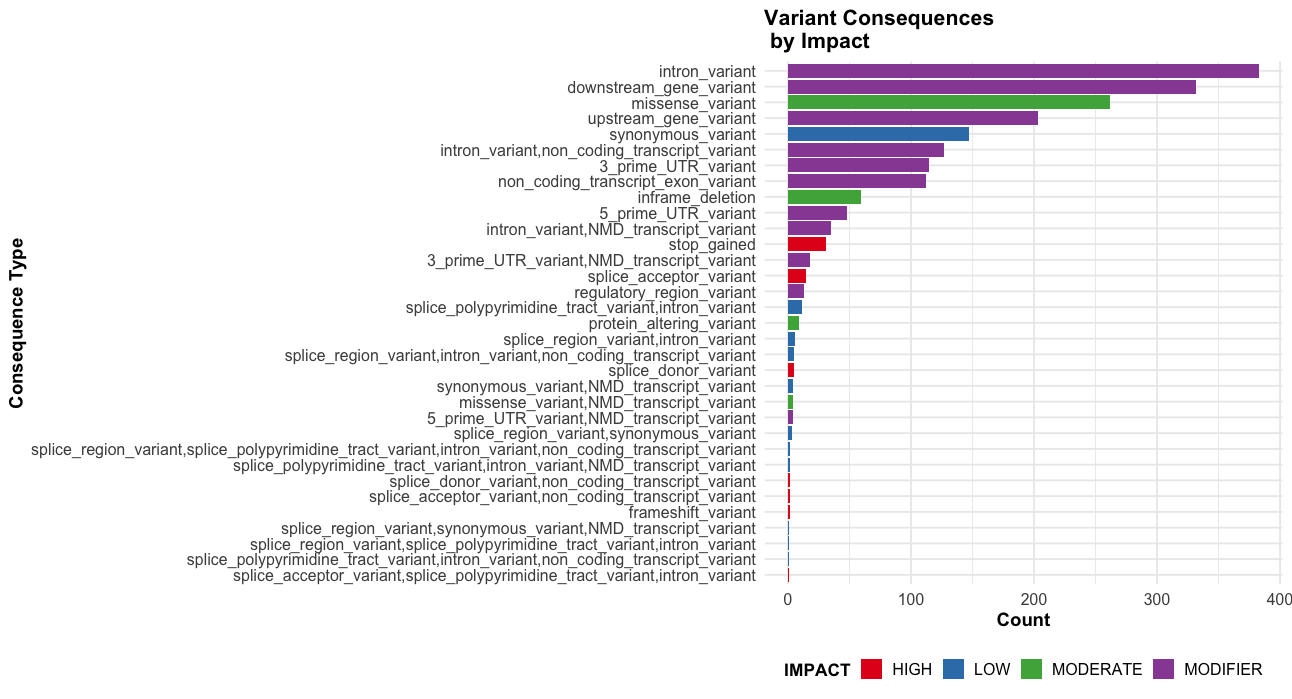
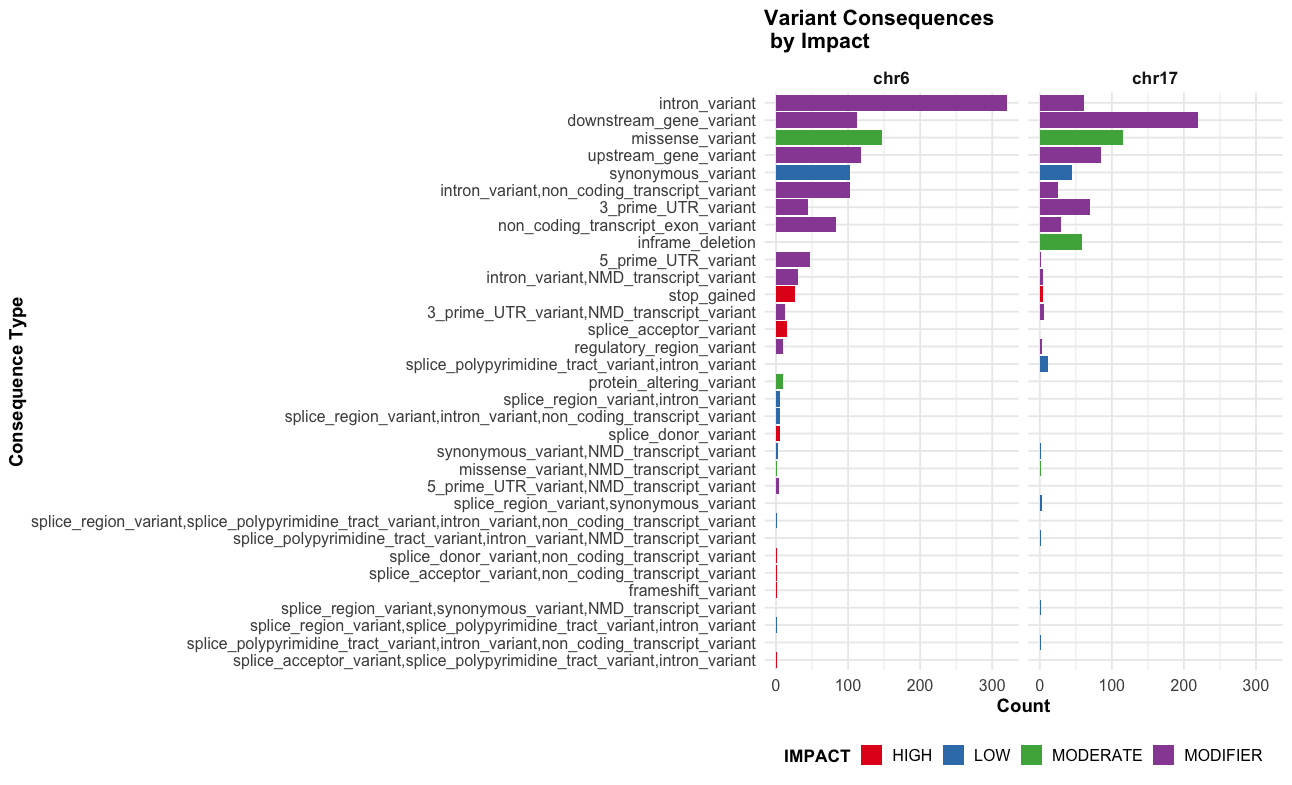
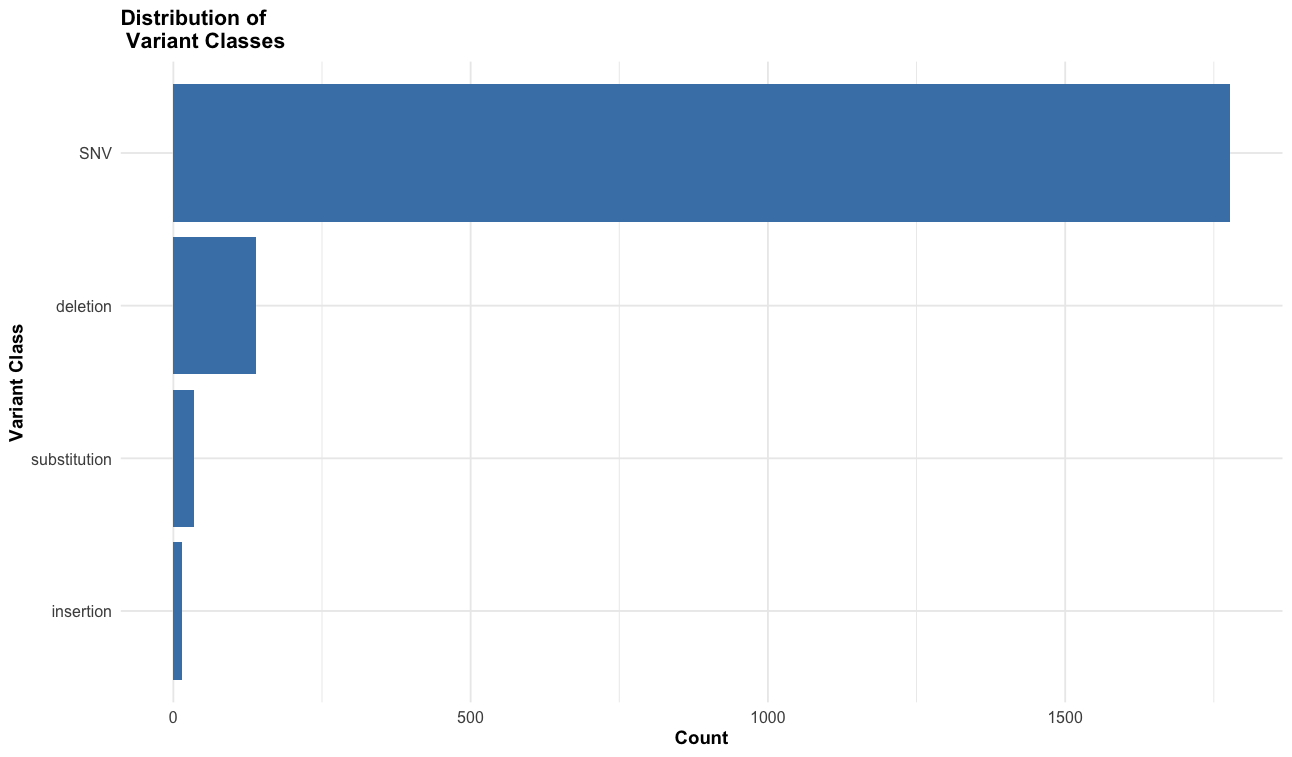
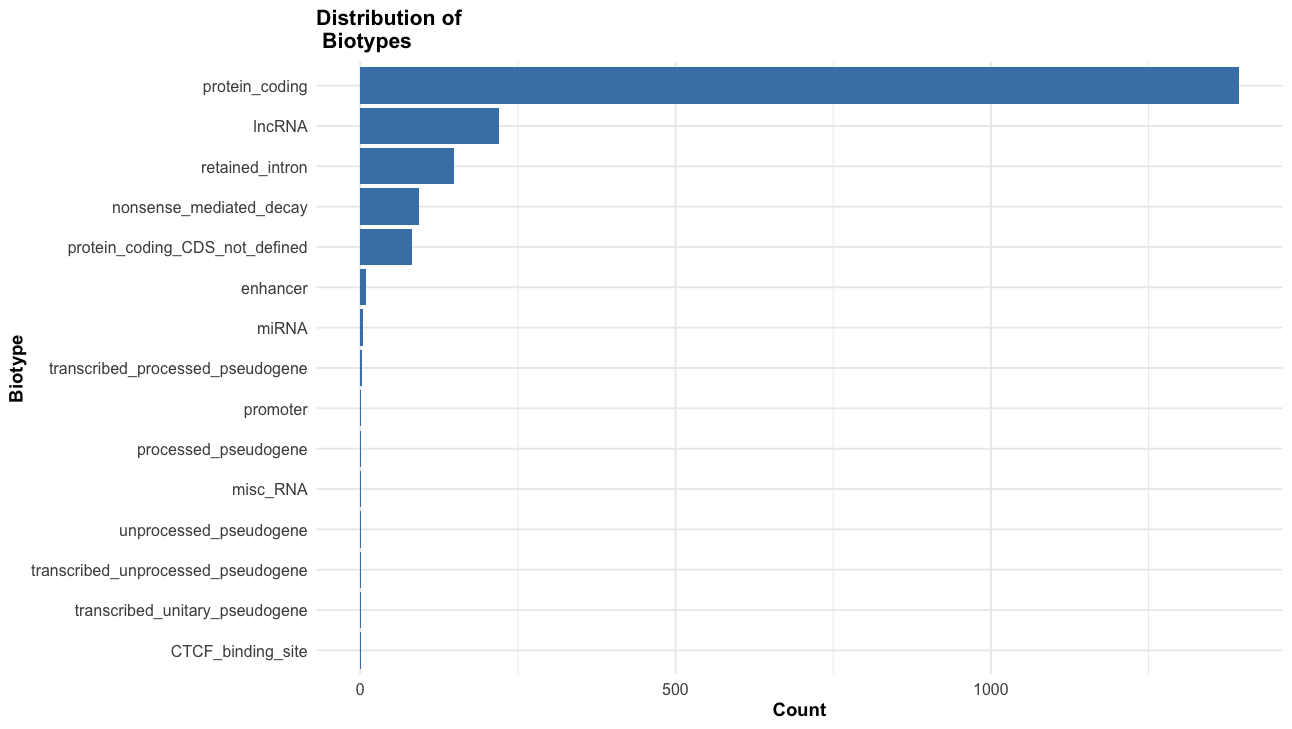
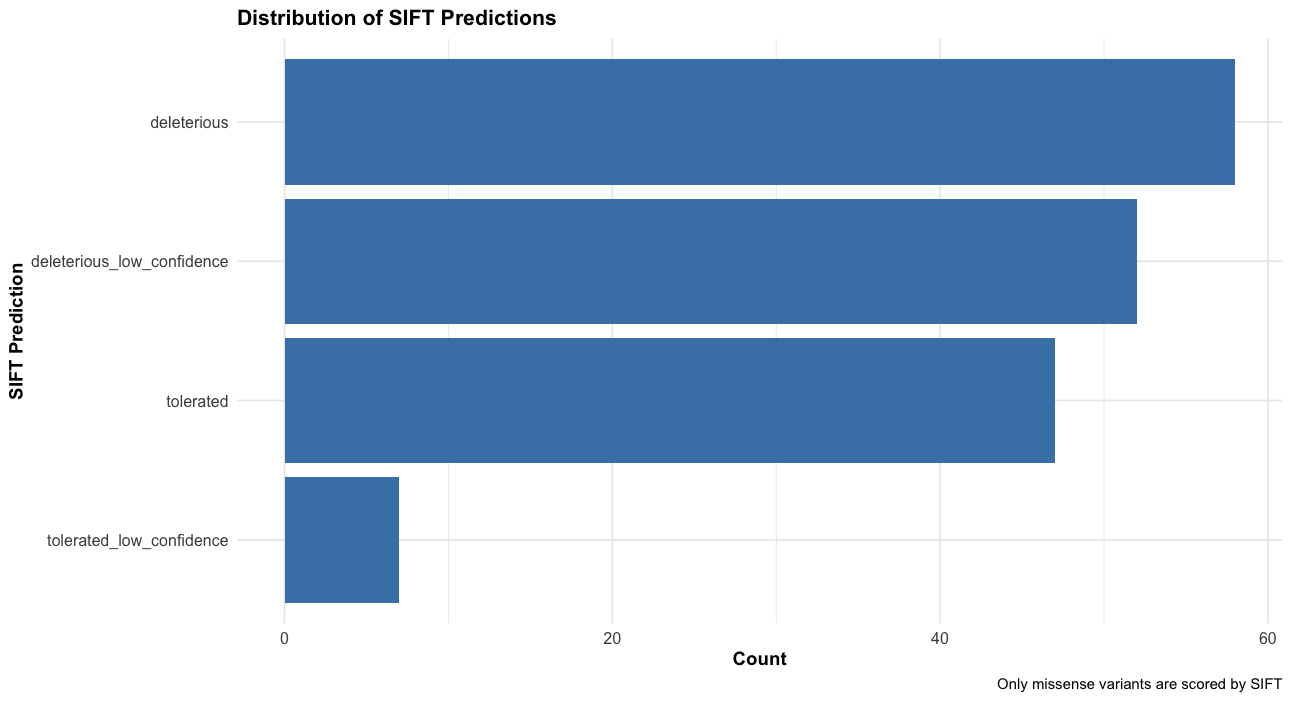
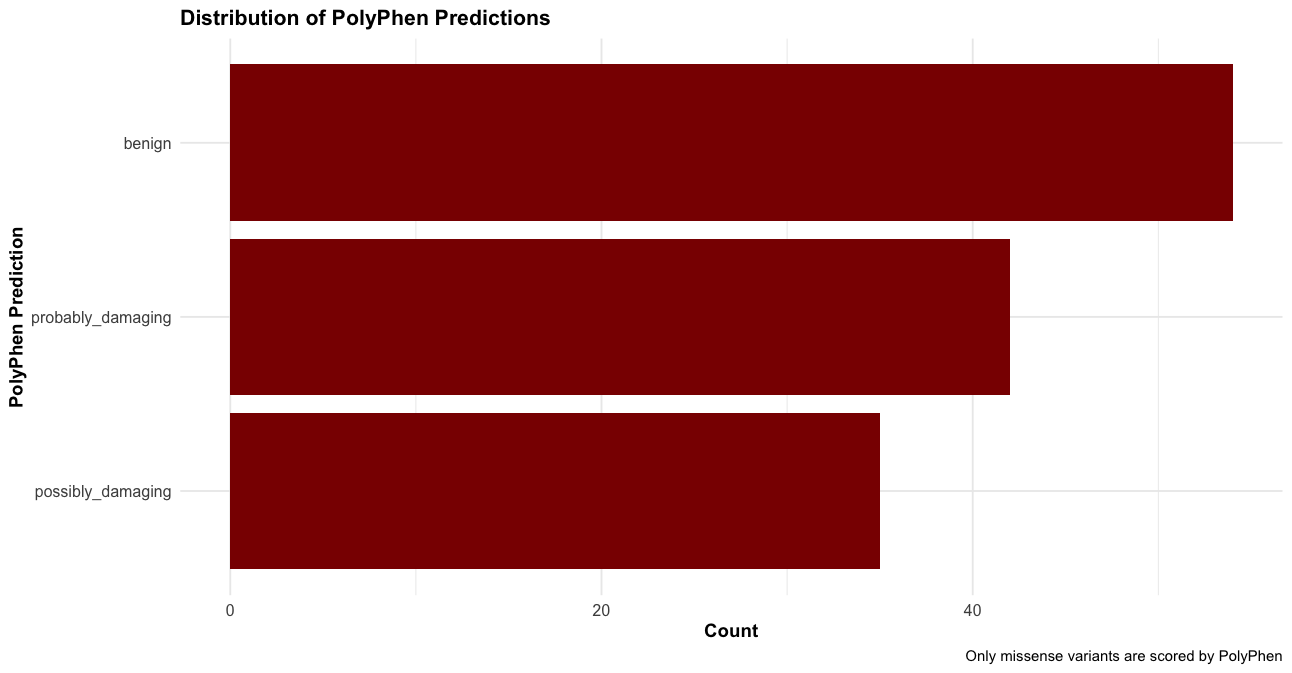
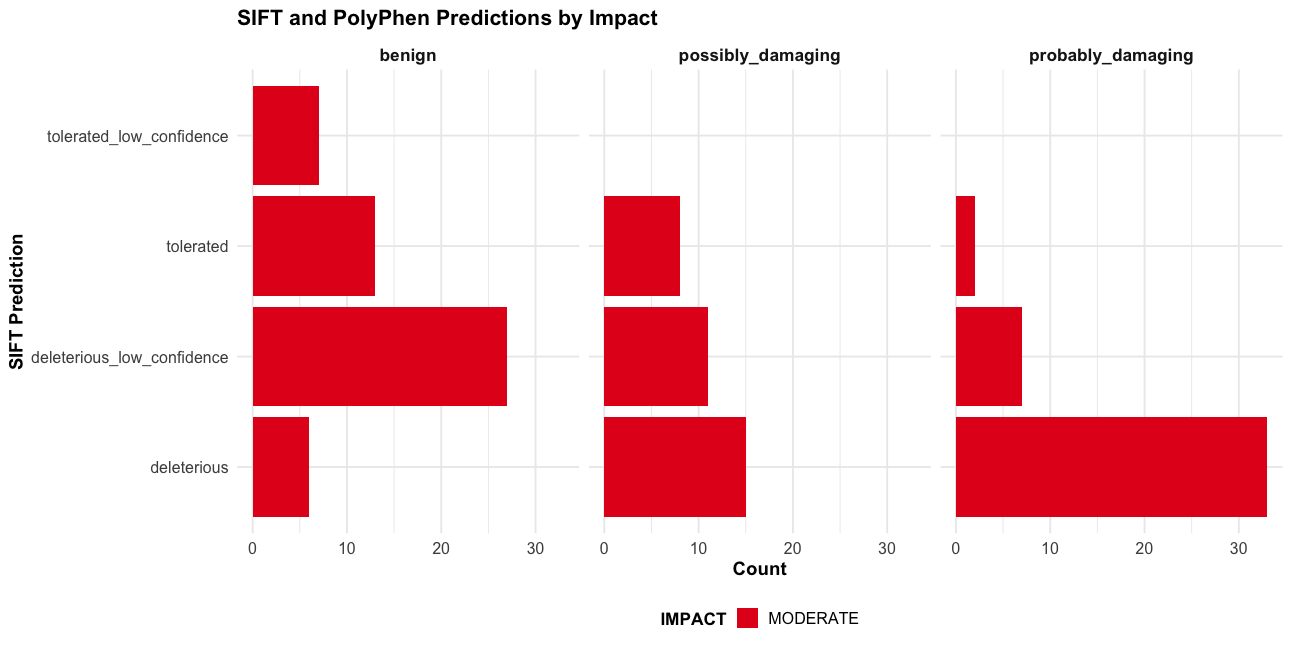
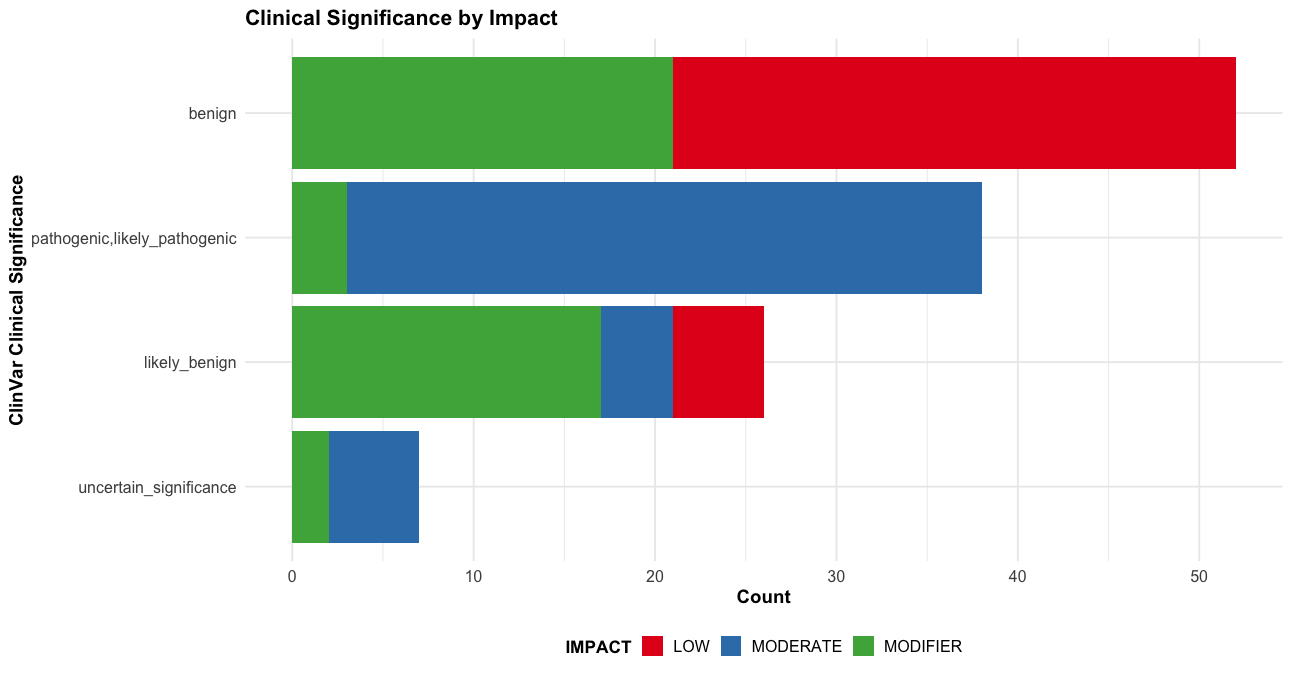
--sift b, --polyphen b, --ccds, --hgvs, --symbol, --numbers, --domains, --regulatory, --canonical, --protein, --biotype, --af, --af_1kg, --af_esp, --af_gnomade, --af_gnomadg, --max_af, --pubmed, --uniprot, --mane, --tsl, --appris, --variant_class, --gene_phenotype, --mirna
This an example of the highput from the TP53 mutation
| Uploaded Variation |
rs28934574 |
dbSNP identifier |
| Location |
chr17:7674894 |
Genomic coordinates (GRCh38) |
| Allele |
A |
Alternative allele |
| Gene |
ENSG00000141510 |
Ensembl gene ID for TP53 |
| Transcript |
ENST00000455263 |
Ensembl transcript ID |
| Feature Type |
Transcript |
Type of genomic feature affected |
| Consequence |
stop_gained |
Creates premature stop codon |
| cDNA Position |
770 |
Position in the cDNA |
| CDS Position |
637 |
Position in the coding sequence |
| Protein Position |
213 |
Position in the protein |
| Amino Acids |
R/* |
Change from Arginine to stop codon |
| Codons |
Cga/Tga |
Codon change |
| Impact |
HIGH |
Severity of variant impact |
| Symbol |
TP53 |
HGNC gene symbol |
| Biotype |
protein_coding |
Type of transcript |
| CCDS |
CCDS45605.1 |
Consensus CDS identifier |
| UniProt |
P04637.305 |
UniProt protein identifier |
| HGVSc |
ENST00000455263.6:c.637C>T |
Coding sequence change in HGVS notation |
| HGVSp |
ENSP00000398846.2:p.Arg213Ter |
Protein change in HGVS notation |
| Clinical Significance |
pathogenic |
Clinical interpretation |
| gnomAD AF |
1.368e-06 |
Allele frequency in gnomAD |
| Maximum AF |
1.656e-05 |
Maximum allele frequency observed |
 Figure 1: Image from the
Figure 1: Image from the