library(SummarizedExperiment)
library(EnrichedHeatmap)
library(gUtils)
library(rtracklayer)
library(circlize)Exercise 3
ChIP and RNA sequencing data plots
Learning Objectives
By the end of this exercise, you will be able to:
- Load and explore
SummarizedExperimentobjects for ChIP-seq and RNA-seq data. - Visualize ChIP-seq signal enrichment across genomic regions using the
EnrichedHeatmappackage. - Generate and interpret RNA-seq expression heatmaps using the
ComplexHeatmappackage. - Transform RNA-seq data to highlight differential expression using log fold-change.
- Annotate heatmaps with sample metadata to enhance interpretability.
- Split and visualize RNA-seq data based on gene regulation status (up- or down-regulated).
- Compare and reflect on the similarities and differences in visualization techniques between ChIP-seq and RNA-seq data.
Load Libraries
ChIP-seq SE object
Here we load the SummarizedExperiment for the ChIP-seq data
Question 1
Similar to ATAC-seq data, please create EnrichedHeatmap for all ChIP-seq datasets and observe difference in genome activity at different genomic regions.
RNA-seq data
rna <- readRDS("data/rna_se.rds")Heatmap of counts
For RNA-seq data, we use ComplexHeatmap package to make heatmaps
Heatmap(matrix = assay(rna, "logCPM"),
name = "RNA",
cluster_columns = FALSE,
show_row_names = FALSE
)Warning: The input is a data frame-like object, convert it to a matrix.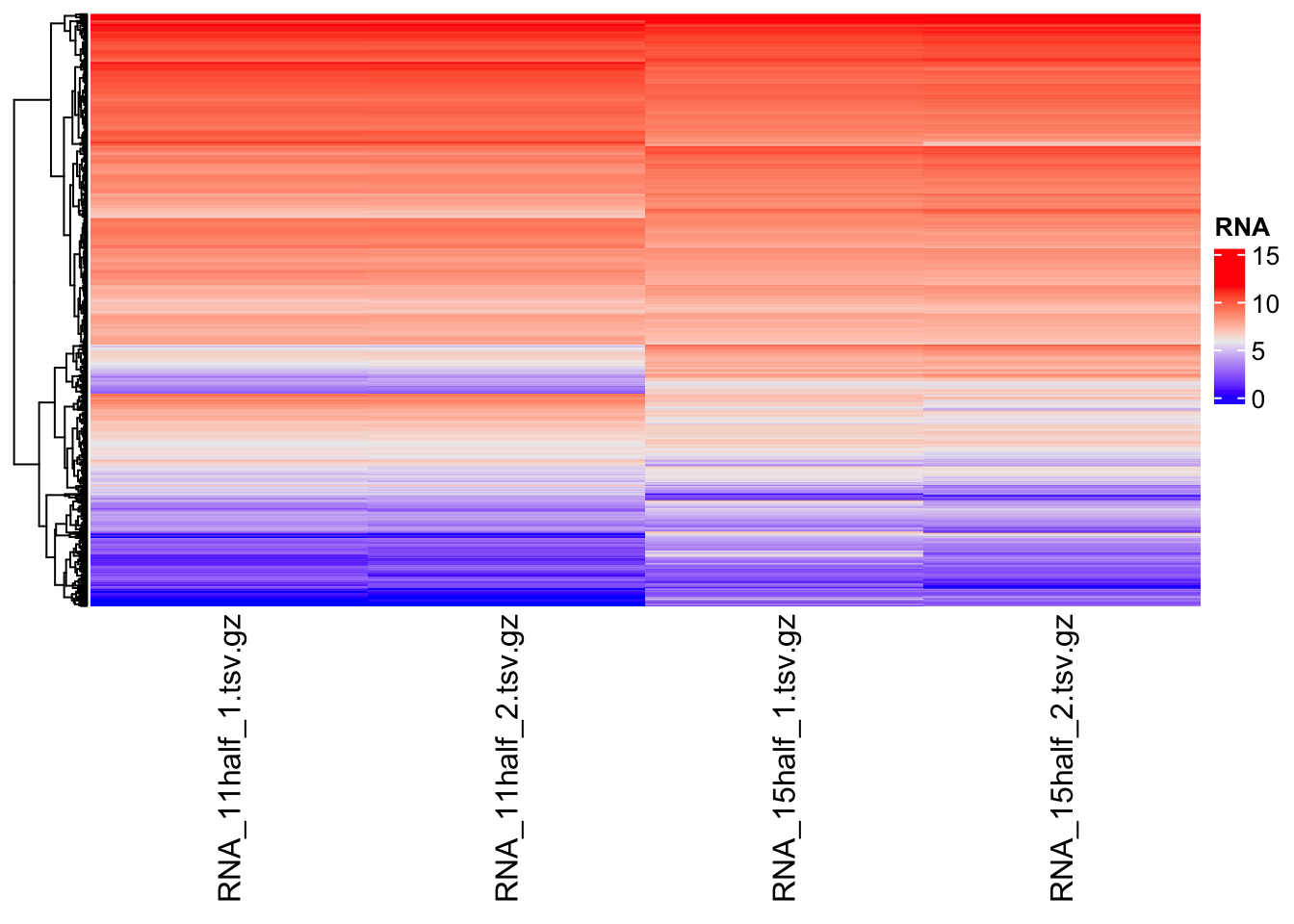
Changing visualization to fold change
Heatmap(matrix = counts,
name = "RNA",
cluster_columns = FALSE,
show_row_names = FALSE
)Warning: The input is a data frame-like object, convert it to a matrix.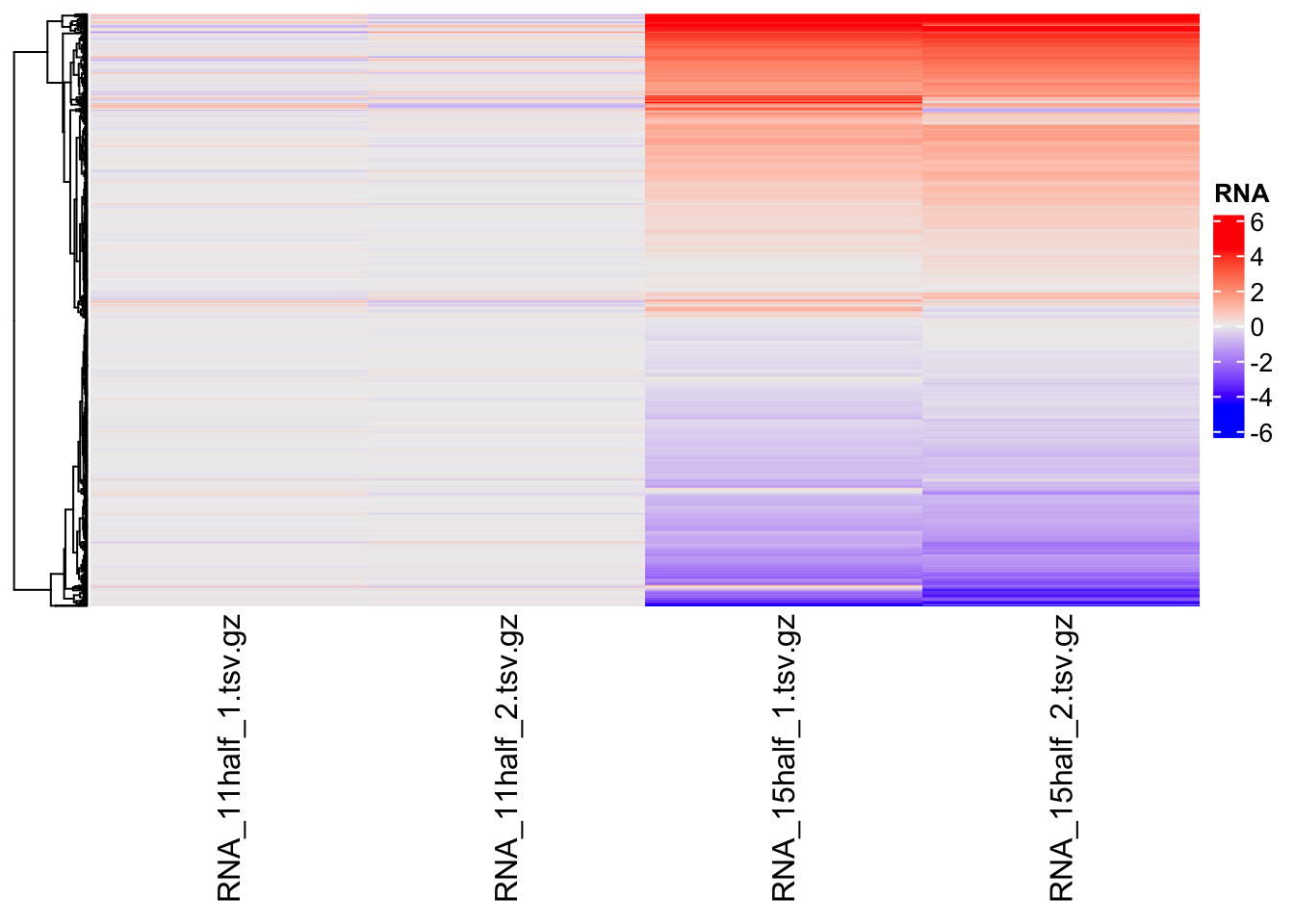
It is now more clear, which genes are up-regulated and which are down-regulated.
Adding colum annotations
colData(rna)DataFrame with 4 rows and 2 columns
sample group
<character> <factor>
RNA_11half_1.tsv.gz RNA_11half_1.tsv.gz 11half
RNA_11half_2.tsv.gz RNA_11half_2.tsv.gz 11half
RNA_15half_1.tsv.gz RNA_15half_1.tsv.gz 15half
RNA_15half_2.tsv.gz RNA_15half_2.tsv.gz 15halfWe can add groups information to the heatmap
Heatmap(matrix = counts,
name = "RNA",
cluster_columns = FALSE,
show_row_names = FALSE,
show_column_names = FALSE,
top_annotation = HeatmapAnnotation(df = colData(rna)[,2,drop = FALSE]),
width = unit(4, "cm"),
height = unit(8, "cm"),
heatmap_legend_param = list(
at = scales::pretty_breaks(n = 3)(range(counts, na.rm = TRUE)),
title = "RNA"
)
)Warning: The input is a data frame-like object, convert it to a matrix.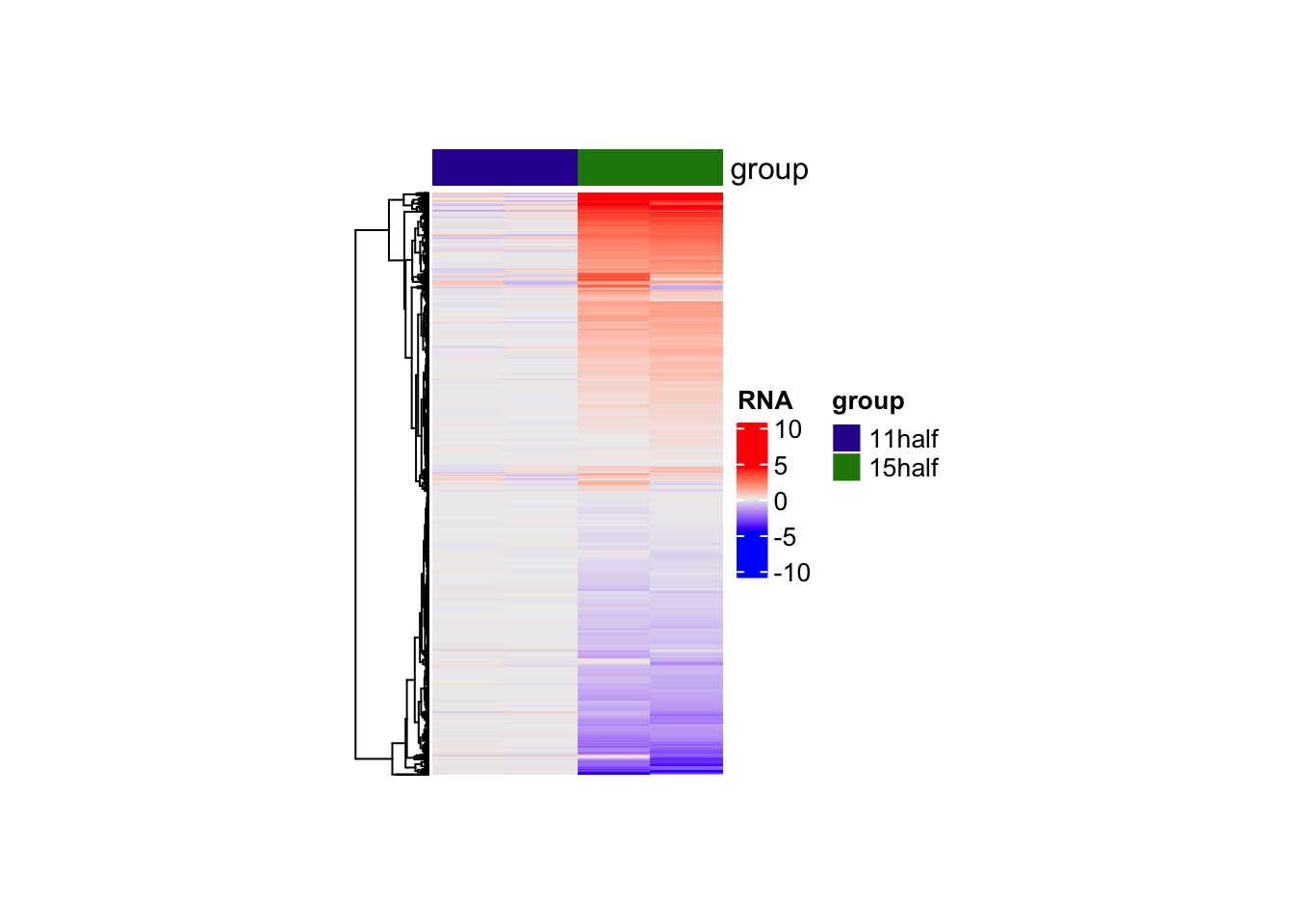
Question 2
Similar to EnrichedHeatmap, we can also split RNA-seq data based on logFC. Can you try to do this?
Tip
logFC <- rowData(rna)[,"logFC"]
names(logFC) <- rownames(rowData(rna))
logFC <- ifelse(logFC > 0, "Up regulated", "Down regulated")
Heatmap(matrix = counts,
name = "RNA",
cluster_columns = FALSE,
show_row_names = FALSE,
show_column_names = FALSE,
top_annotation = HeatmapAnnotation(df = colData(rna)[,2,drop = FALSE]),
width = unit(4, "cm"),
height = unit(8, "cm"),
heatmap_legend_param = list(
at = scales::pretty_breaks(n = 3)(range(counts, na.rm = TRUE)),
title = "RNA"
),
row_split = logFC
)Warning: The input is a data frame-like object, convert it to a matrix.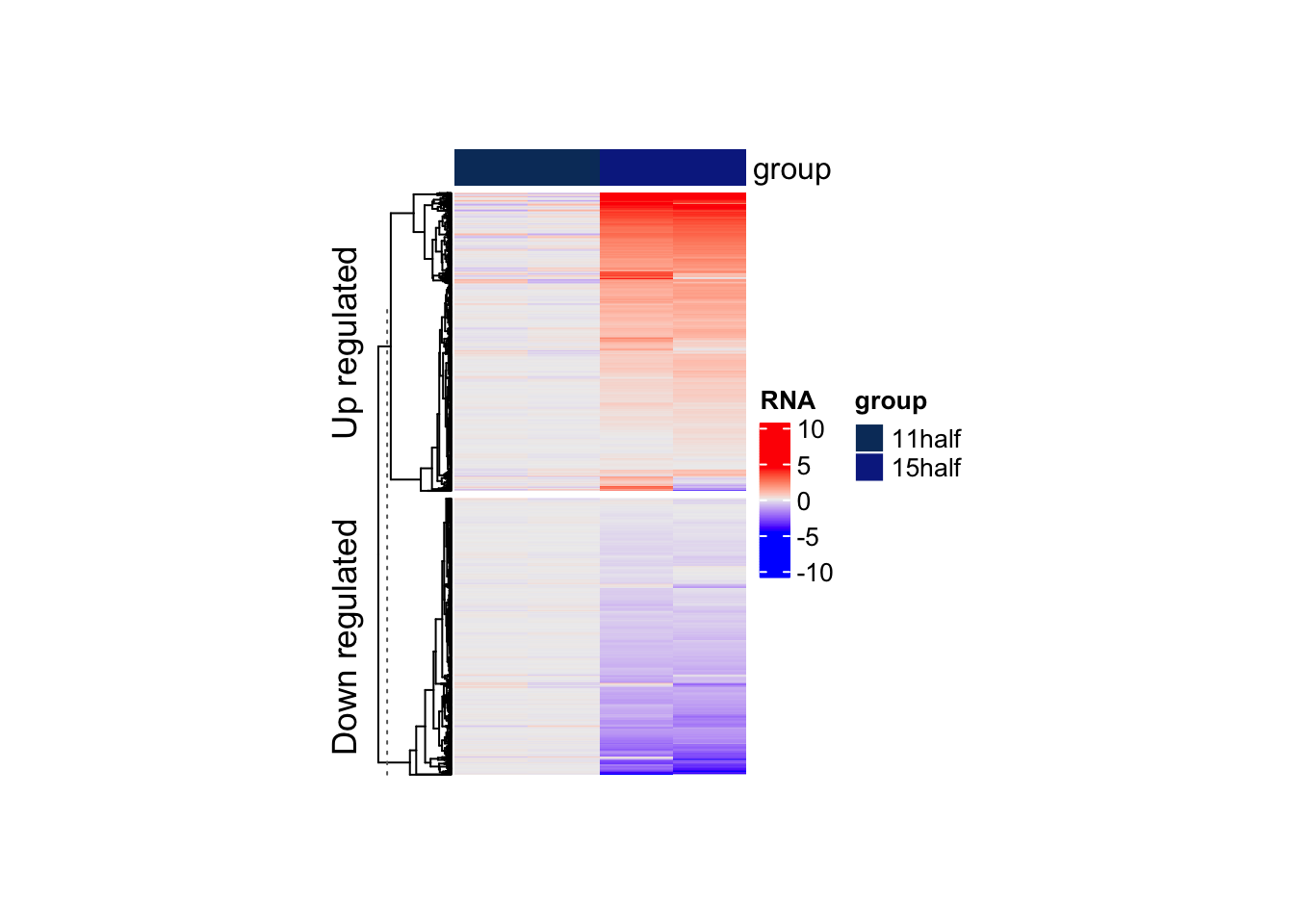
Important
Heatmap and EnrichedHeatmap have a lot of options in common.