Decorating and optimising a Snakemake workflow
Learning outcomes
After having completed this chapter you will be able to:
- Optimise a workflow by multi-threading
- Use non-file parameters and config files in rules
- Create rules with non-conventional outputs
- Modularise a workflow
- Make a workflow process a list of files rather than one file at a time
Exercises
In this series of exercises, we will create only one new rule to add to our workflow, because this part aims mainly to show how to improve and ‘decorate’ the rules we previously wrote.
Development and back-up
During this session, we will modify our Snakefile quite heavily, so it may be a good idea to start by making a back-up: cp worklow/Snakefile worklow/Snakefile_backup. As a general rule, if you have a doubt on the code you are developing, do not hesitate to make a back-up.
Optimising a workflow by multi-threading
When working with real datasets, most processes are very long and computationally expensive. Fortunately, they can be parallelised very efficiently to decrease the computation time by using several threads for a single job.
Exercise: Parallelise as much processes as possible using the threads directive and test its effect:
- Identify which software can make use of parallelisation
- Identify in each software the parameter that controls multi-threading
- Implement the multi-threading
Hint
- Check the software documentation and parameters with the
-h/--helpflags - Remember that multi-threading only applies to software that can make use of a threads parameters, Snakemake itself cannot parallelise a software automatically
- Remember that you need to add threads to the Snakemake rule but also to the commands! Just increasing the number of threads in Snakemake will not magically run a command with multiple threads
- Remember that you have 4 threads in total, so even if you ask for more in a rule, Snakemake will cap this value at 4. And if you use 4 threads in a rule, that means that no other job can run parallel!
Answer
It turns out that all the software except samtools index can handle multi-threading:
atropos trim,hisat2,samtools view, andsamtools sortuse the--threadsoptionfeatureCountsuses the-Toption
Let’s use 4 threads for the mapping step and 2 for the other steps. Your Snakefile should look like this:
rule fastq_trim:
'''
This rule trims paired-end reads to improve their quality. Specifically, it removes:
- Low quality bases
- A stretches longer than 20 bases
- N stretches
'''
input:
reads1 = 'data/{sample}_1.fastq',
reads2 = 'data/{sample}_2.fastq',
output:
trim1 = 'results/{sample}/{sample}_atropos_trimmed_1.fastq',
trim2 = 'results/{sample}/{sample}_atropos_trimmed_2.fastq'
log:
'logs/{sample}/{sample}_atropos_trimming.log'
benchmark:
'benchmarks/{sample}/{sample}_atropos_trimming.txt'
resources:
mem_mb = 500
threads: 2
shell:
'''
echo "Trimming reads in <{input.reads1}> and <{input.reads2}>" > {log}
atropos trim -q 20,20 --minimum-length 25 --trim-n --preserve-order --max-n 10 \
--no-cache-adapters -a "A{{20}}" -A "A{{20}}" --threads {threads} \
-pe1 {input.reads1} -pe2 {input.reads2} -o {output.trim1} -p {output.trim2} &>> {log}
echo "Trimmed files saved in <{output.trim1}> and <{output.trim2}> respectively" >> {log}
echo "Trimming report saved in <{log}>" >> {log}
'''
rule read_mapping:
'''
This rule maps trimmed reads of a fastq on a reference assembly.
'''
input:
trim1 = rules.fastq_trim.output.trim1,
trim2 = rules.fastq_trim.output.trim2
output:
sam = 'results/{sample}/{sample}_mapped_reads.sam',
report = 'results/{sample}/{sample}_mapping_report.txt'
log:
'logs/{sample}/{sample}_mapping.log'
benchmark:
'benchmarks/{sample}/{sample}_mapping.txt'
resources:
mem_gb = 2
threads: 4
shell:
'''
echo "Mapping the reads" > {log}
hisat2 --dta --fr --no-mixed --no-discordant --time --new-summary --no-unal \
-x resources/genome_indices/Scerevisiae_index --threads {threads} \
-1 {input.trim1} -2 {input.trim2} -S {output.sam} --summary-file {output.report} 2>> {log}
echo "Mapped reads saved in <{output.sam}>" >> {log}
echo "Mapping report saved in <{output.report}>" >> {log}
'''
rule sam_to_bam:
'''
This rule converts a sam file to bam format, sorts it and indexes it.
'''
input:
sam = rules.read_mapping.output.sam
output:
bam = 'results/{sample}/{sample}_mapped_reads.bam',
bam_sorted = 'results/{sample}/{sample}_mapped_reads_sorted.bam',
index = 'results/{sample}/{sample}_mapped_reads_sorted.bam.bai'
log:
'logs/{sample}/{sample}_mapping_sam_to_bam.log'
benchmark:
'benchmarks/{sample}/{sample}_mapping_sam_to_bam.txt'
resources:
mem_mb = 250
threads: 2
shell:
'''
echo "Converting <{input.sam}> to BAM format" > {log}
samtools view {input.sam} --threads {threads} -b -o {output.bam} 2>> {log}
echo "Sorting BAM file" >> {log}
samtools sort {output.bam} --threads {threads} -O bam -o {output.bam_sorted} 2>> {log}
echo "Indexing the sorted BAM file" >> {log}
samtools index -b {output.bam_sorted} -o {output.index} 2>> {log}
echo "Sorted file saved in <{output.bam_sorted}>" >> {log}
'''
rule reads_quantification_genes:
'''
This rule quantifies the reads of a bam file mapping on genes and produces
a count table for all genes of the assembly. The strandedness parameter
is determined by get_strandedness().
'''
input:
bam_once_sorted = rules.sam_to_bam.output.bam_sorted,
output:
gene_level = 'results/{sample}/{sample}_genes_read_quantification.tsv',
gene_summary = 'results/{sample}/{sample}_genes_read_quantification.summary'
log:
'logs/{sample}/{sample}_genes_read_quantification.log'
benchmark:
'benchmarks/{sample}/{sample}_genes_read_quantification.txt'
resources:
mem_mb = 500
threads: 2
shell:
'''
echo "Counting reads mapping on genes in <{input.bam_once_sorted}>" > {log}
featureCounts -t exon -g gene_id -s 2 -p -B -C --largestOverlap --verbose -F GTF \
-a resources/Scerevisiae.gtf -T {threads} -o {output.gene_level} {input.bam_once_sorted} &>> {log}
echo "Renaming output files" >> {log}
mv {output.gene_level}.summary {output.gene_summary}
echo "Results saved in <{output.gene_level}>" >> {log}
echo "Report saved in <{output.gene_summary}>" >> {log}
'''
Exercise: Finally, test the effect of the number of threads on the workflow’s runtime. What command will you use to run the workflow? Does the workflow run faster?
Answer
The command to use is:
snakemake --cores 4 -F -r -p results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv
Do not forget to provide additional cores to Snakemake in the execution command with --cores 4. Note that the number of threads allocated to all jobs running at a given time cannot exceed the value specified with --cores. Therefore, if you leave this number at 1, Snakemake will not be able to use multiple threads. Also note that increasing --cores allows Snakemake to run multiple jobs in parallel (for example, running 2 jobs using 2 threads each). The workflow now takes ~6 min to run, compared to ~10 min before (i.e. a 40% decrease!). This gives you an idea of how powerful multi-threading is when the datasets and computing power get bigger!
Explicit is better than implicit
Even if a software cannot multi-thread, it is useful to add threads: 1 in the rule to keep the rule consistency and clearly state that the software works with a single thread.
Things to keep in mind when using parallel execution
- Parallel jobs will use more RAM. If you run out then either your OS will swap data to disk, or a process will crash
- The on-screen output from parallel jobs will be mixed, so save any output to log files instead
Using non-file parameters and config files
Non-file parameters
As we have seen, Snakemake’s execution is based around inputs and outputs of each step of the workflow. However, a lot of software rely on additional non-file parameters. In the previous presentation and series of exercises, we advocated (rightfully so!) against using hard-coded filepaths. Yet, if you look back at the rules we have implemented, you will find 2 occurrences of this behaviour in the shell command:
- In the
rule read_mapping, the index parameter-x resources/genome_indices/Scerevisiae_index - In the
rule reads_quantification_genes, the annotation parameter-a resources/Scerevisiae.gtf
This reduces readability and also makes it very hard to change the value of these parameters.
The params directive was designed for this purpose: it allows to specify additional parameters that can also depend on the wildcard values and use input functions (see Session 4 for more information on this). params values can be of any type (integer, string, list etc…) and similarly to the {input} and {output} placeholders, they can also be accessed from the shell command with the placeholder {params}. Just like for the input and output directives, you can define multiple parameters (in this case, do not forget the comma between each entry!) and they can be named (in practice, unknown parameters are unexplicit and easily confusing, so parameters should always be named!).
It also helps readability and clarity to use the params section to name and assign parameters and variables for your shell command.
Here is an example on how to use params:
rule example:
input:
'data/example.tsv'
output:
'results/example.txt'
params:
lines = 5
shell:
'head -n {params.lines} {input} > {output}'
Parameters arguments
In contrast to the input directive, the params directive can optionally take more arguments than only wildcards, namely input, output, threads, and resources.
Exercise: Replace the two hard-coded paths mentioned earlier by params.
Hint
Add a params directive to the rules, name the parameter and replace the path by the placeholder in the shell command.
Answer
Note: for clarity, only the lines that changed are shown below.
rule read_mapping
params:
index = 'resources/genome_indices/Scerevisiae_index'
shell:
'hisat2 --dta --fr --no-mixed --no-discordant --time --new-summary --no-unal \
-x {params.index} --threads {threads} \
-1 {input.trim1} -2 {input.trim2} -S {output.sam} --summary-file {output.report} 2>> {log}'
rule reads_quantification_genes
params:
annotations = 'resources/Scerevisiae.gtf'
shell:
'featureCounts -t exon -g gene_id -s 2 -p -B -C --largestOverlap --verbose -F GTF \
-a {params.annotations} -T {threads} -o {output.gene_level} {input.bam_once_sorted} &>> {log}'
Snakemake re-run behaviour
If you try to re-run only the last rule with snakemake --cores 4 -r -p -f results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv, Snakemake will actually try to re-run 3 rules in total.
This is because the code changed in 2 rules (see reason field in Snakemake’s log), which triggered an update of the inputs in the 3rd rule (sam_to_bam). To avoid this, first touch the files with snakemake --cores 1 --touch -F results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv then re-run the last rule.
Config files
That being said, there is an even better way to handle parameters like the we just modified: instead of hard-coding parameter values in the Snakefile, Snakemake allows to define parameters and their values in config files. The config files will be parsed by Snakemake when executing the workflow, and parameters and their values will be stored in a Python dictionary named config. The path to the config file can be specified either in the Snakefile with the line configfile: <path/to/file.yaml> at the top of the file, or it can be specified at runtime with the execution parameter --configfile <path/to/file.yaml>.
Config files are stored in the config subfolder and written in the JSON or YAML format. We will use the latter for this course as it is the most user-friendly and the recommended one. Briefly, in the YAML format, parameters are defined with the syntax <name>: <value>. Values can be strings, integers, floating points, booleans … For a complete overview of available value types, see this list. A parameter can have multiple values, which are then each listed on an indented single line starting with “-”. These values will be stored in a Python list when Snakemake parses the config file. Finally, parameters can be nested on indented single lines, and they will be stored as a dictionary when Snakemake parses the config file.
The example below shows a parameter with a single value (lines_number), a parameter with multiple values (samples), and an example of nested parameters (resources):
# Parameter with a single value (string, int, float, bool ...)
lines_number: 5
# Parameter with multiple values
samples:
- sample1
- sample2
# Nested parameters
resources:
threads: 4
memory: 4G
Then, each parameter can be accessed in Snakefile with the following syntax:
config['lines_number'] # --> 5
config['samples'] # --> ['sample1', 'sample2'] # Lists of parameters become list
config['resources'] # --> {'threads': 4, 'memory': '4G'} # Lists of named parameters become dictionaries
config['resources']['threads'] # --> 4
Accessing config values in shell
Values stored in the config dictionary cannot be accessed directly within the shell directive. If you need to use a parameter value in shell, define the parameter in params and assign its value from the config dictionary.
Exercise: Create a config file in YAML format and fill it with adapted variables and values to replace the 2 hard-coded parameters in rules read_mapping and reads_quantification_genes. Then replace the hard-coded parameters by values from the config file and add its path on top of your Snakefile.
Answer
Note: for clarity, only the lines that changed are shown below. The first step is to create the subfolder and an empty config file:
mkdir config # Create a new folder
touch config/config.yaml # Create an empty config file
Then, fill the config file with the desired values:
# Configuration options of RNAseq-analysis workflow
# Location of the genome indices
index: 'resources/genome_indices/Scerevisiae_index'
# Location of the annotation file
annotations: 'resources/Scerevisiae.gtf'
Then, replace the params values in the Snakefile:
rule read_mapping
params:
index = config['index']
rule reads_quantification_genes
params:
annotations = config['annotations']
Finally, add the file path on top of the Snakefile:
configfile: 'config/config.yaml'
Now, if we need to change these values, we can easily do it in the config file instead of modifying the code!
Using non-conventional outputs
Snakemake has several built-in utilities to assign properties to outputs that are deemed ‘special’. These properties are listed in the table below:
| Property | Syntax | Function |
|---|---|---|
| Temporary | temp('path/to/file.txt') |
File is deleted as soon as it is not required by any future jobs |
| Protected | protected('path/to/file.txt') |
File cannot be overwritten after the job ends (useful to prevent erasing a file by mistake) |
| Ancient | ancient('path/to/file.txt') |
File will not be re-created when running the pipeline (useful for files that require heavy computation) |
| Directory | directory('path/to/directory') |
Output is a directory instead of a file (use ‘touch’ instead if possible) |
| Touch | touch('path/to/file.txt') |
Create an empty flag file ‘file.txt’ regardless of the shell command (if the command finished without errors) |
The next paragraphs will show how to use some of these properties.
Use-case of the temp() command
Exercise: Can you think of a convenient use of temp() command?
Answer
The temp() command is extremely useful to automatically remove intermediary outputs that are no longer needed.
Exercise: In your workflow, identify outputs that are intermediary and mark them as temporary with temp().
Answer
The unsorted .bam and the .sam outputs seem like great candidates to be marked as temporary. One could also argue that the trimmed FASTQ files are also temporary, but we will keep them for now. Note: for clarity, only the lines that changed are shown below.
rule read_mapping
output:
sam = temp('results/{sample}/{sample}_mapped_reads.sam'),
rule sam_to_bam
output:
bam = temp('results/{sample}/{sample}_mapped_reads.bam'),
Consequences of using temp()
Removing temporary outputs is a great way to save a lot of storage space. If you look at the size of your current results/ folder (du -bchd0 results/), you will notice that it drastically. Just removing these two files would allow to save ~1 GB. While it may not seem a lot, remember that you usually have much bigger files and many more samples! On the other hand, using temporary outputs might force you to re-run more jobs than necessary if an input changes, so carefully think about it before using it.
Exercise: On the contrary, is there a file of your workflow that you would like to protect with protected()
Answer
This is debatable, but one could argue that the sorted .bam file is a good candidate for protection.
rule sam_to_bam
output:
bam_sorted = protected('results/{sample}/{sample}_mapped_reads_sorted.bam'),
If you set this output as protected, be careful when you want to re-run your workflow and recreate the file!
Use-case of the directory() command: the FastQC example
FastQC is a program designed to spot potential problems in high-througput sequencing datasets. It is a very popular tool, notably because it runs quickly and does not require a lot of configuration. It runs a set of analyses on one or more raw sequence files in FASTQ or BAM format and produces a report with quality plots that summarises the results. It will highlight any areas where a dataset looks unusual and might require a closer look. As such, it would be interesting to run FastQC on the original .fastq files and the trimmed .fastq files to check whether trimminga actually improved the read quality. FastQC can be run interactively or in batch mode, during which it saves results as an HTML file and a ZIP file. We will soon see that running FastQC in batch mode presents a little problem.
Data types and FastQC
FastQC does not differentiate between sequencing techniques and as such can be used to look at libraries coming from a large number of experiments (Genomic Sequencing, ChIP-Seq, RNAseq, BS-Seq etc…).
If you run fastqc -h, you will notice something a bit surprising (but not unusual in bioinformatics):
-o --outdir Create all output files in the specified output directory.
Please note that this directory must exist as the program
will not create it. If this option is not set then the
output file for each sequence file is created in the same
directory as the sequence file which was processed.
-d --dir Selects a directory to be used for temporary files written when
generating report images. Defaults to system temp directory if
not specified.
Two files are produced for each FASTQ file and these files appear in the same directory as the input file: FastQC does not allow to specify the names of the output files! However, we can set an alternative output directory, even though it needs to be created before FastQC is run.
There are different solutions to deal with this problem:
- Work with the default file names produced by FastQC and leave the reports in the same directory than the input files
- Create the outputs in a new directory and leave the reports with their default name
- Create the outputs in a new directory and tell Snakemake that the directory itself is the output
- Force a naming convention by renaming the FastQC output files within the rule
For the sake of time, we will not test all 4 solutions, but rather try to apply the 3rd or the 4th solution. We’ll briefly summarise solutions 1 and 2 here:
- This could work, but it’s better not to put the reports in the same directory than the input sequences. As a general principle, when writing Snakemake rules, we prefer to be in charge of the output names and to have all the files linked to a sample in the same directory
- This involves manually constructing the output directory path to use with the
-ooption, which works but isn’t very convenient
The base of the FastQC command is the following: fastqc --format fastq --threads 2 <input_fastq1> <input_fastq2>
-t/--threads: specify the number of files which can be processed simultaneously. Here, it will be 2 because the inputs are paired-end files- The
-oand-dwill be used in the last 2 solutions that we will now see in details - We will create a single rule to run FastQC on both the original and the trimmed FASTQ files
Choose only one solution to implement:
This option amounts to tell Snakemake not to worry about individual files at all and consider the output of the rule as an entire directory.
Exercise: Implement a single rule to run FastQC on both the original and the trimmed FASTQ files (4 files in total) using directories as ouputs with the directory() command.
Answer
This makes the rule definition quite ‘simple’:
rule fastq_qc_sol3:
'''
This rule performs a QC on paired-end fastq files before and after trimming.
'''
input:
reads1 = rules.fastq_trim.input.reads1,
reads2 = rules.fastq_trim.input.reads2,
trim1 = rules.fastq_trim.output.trim1,
trim2 = rules.fastq_trim.output.trim2
output:
before_trim = directory('results/{sample}/fastqc_reports/before_trim/'),
after_trim = directory('results/{sample}/fastqc_reports/after_trim/')
log:
'logs/{sample}/{sample}_fastqc.log'
benchmark:
'benchmarks/{sample}/{sample}_atropos_fastqc.txt'
resources:
mem_gb = 1
threads: 2
shell:
'''
echo "Creating output directory <{output.before_trim}>" > {log}
mkdir -p {output.before_trim} 2>> {log}
echo "Performing QC of reads before trimming in <{input.reads1}> and <{input.reads2}>" >> {log}
fastqc --format fastq --threads {threads} --outdir {output.before_trim} \
--dir {output.before_trim} {input.reads1} {input.reads2} &>> {log}
echo "Results saved in <{output.before_trim}>" >> {log}
echo "Creating output directory <{output.after_trim}>" >> {log}
mkdir -p {output.after_trim} 2>> {log}
echo "Performing QC of reads after trimming in <{input.trim1}> and <{input.trim2}>" >> {log}
fastqc --format fastq --threads {threads} --outdir {output.after_trim} \
--dir {output.after_trim} {input.trim1} {input.trim2} &>> {log}
echo "Results saved in <{output.after_trim}>" >> {log}
'''
.snakemake_timestamp
When directory() is used, Snakemake creates an empty file called .snakemake_timestamp in the output directory. This is the marker it uses to know if it needs to re-run the rule producing the directory.
Overall, this rule works well and allows for an easy rule definition. However, in this case, individual files are not explicitly named as outputs and this may cause problems to chain rules later. Also, remember that some applications won’t give you any control at all over the outputs, which is why you need a back-up plan, i.e. solution 4: the most powerful solution is to use shell commands to move and/or rename the files to the names you want. Also, the Snakemake developers advise to use directory() as a last resort and to rather use the touch() flag instead.
This option amounts to let FastQC follows its default behaviour but force the renaming of the files afterwards to obtain the exact outputs we require.
Exercise: Implement a single rule to run FastQC on both the original and the trimmed FASTQ files (4 files in total) and rename the files created by FastQC to precise output names using the mv command.
Answer
This makes the rule definition (much) more complicated than the other solution:
rule fastq_qc_sol4:
'''
This rule performs a QC on paired-end fastq files before and after trimming.
'''
input:
reads1 = rules.fastq_trim.input.reads1,
reads2 = rules.fastq_trim.input.reads2,
trim1 = rules.fastq_trim.output.trim1,
trim2 = rules.fastq_trim.output.trim2
output:
# QC before trimming
html1_before = 'results/{sample}/fastqc_reports/{sample}_before_trim_1.html',
zipfile1_before = 'results/{sample}/fastqc_reports/{sample}_before_trim_1.zip',
html2_before = 'results/{sample}/fastqc_reports/{sample}_before_trim_2.html',
zipfile2_before = 'results/{sample}/fastqc_reports/{sample}_before_trim_2.zip',
# QC after trimming
html1_after = 'results/{sample}/fastqc_reports/{sample}_after_trim_1.html',
zipfile1_after = 'results/{sample}/fastqc_reports/{sample}_after_trim_1.zip',
html2_after = 'results/{sample}/fastqc_reports/{sample}_after_trim_2.html',
zipfile2_after = 'results/{sample}/fastqc_reports/{sample}_after_trim_2.zip'
params:
wd = 'results/{sample}/fastqc_reports/',
# QC before trimming
html1_before = 'results/{sample}/fastqc_reports/{sample}_1_fastqc.html',
zipfile1_before = 'results/{sample}/fastqc_reports/{sample}_1_fastqc.zip',
html2_before = 'results/{sample}/fastqc_reports/{sample}_2_fastqc.html',
zipfile2_before = 'results/{sample}/fastqc_reports/{sample}_2_fastqc.zip',
# QC after trimming
html1_after = 'results/{sample}/fastqc_reports/{sample}_atropos_trimmed_1_fastqc.html',
zipfile1_after = 'results/{sample}/fastqc_reports/{sample}_atropos_trimmed_1_fastqc.zip',
html2_after = 'results/{sample}/fastqc_reports/{sample}_atropos_trimmed_2_fastqc.html',
zipfile2_after = 'results/{sample}/fastqc_reports/{sample}_atropos_trimmed_2_fastqc.zip'
log:
'logs/{sample}/{sample}_fastqc.log'
benchmark:
'benchmarks/{sample}/{sample}_atropos_fastqc.txt'
resources:
mem_gb = 1
threads: 2
shell:
'''
echo "Performing QC of reads before trimming in <{input.reads1}> and <{input.reads2}>" >> {log}
fastqc --format fastq --threads {threads} --outdir {params.wd} \
--dir {params.wd} {input.reads1} {input.reads2} &>> {log}
echo "Renaming results from original fastq analysis" >> {log} # Renames files because we can't choose fastqc output
mv {params.html1_before} {output.html1_before} 2>> {log}
mv {params.zipfile1_before} {output.zipfile1_before} 2>> {log}
mv {params.html2_before} {output.html2_before} 2>> {log}
mv {params.zipfile2_before} {output.zipfile2_before} 2>> {log}
echo "Performing QC of reads after trimming in <{input.trim1}> and <{input.trim2}>" >> {log}
fastqc --format fastq --threads {threads} --outdir {params.wd} \
--dir {params.wd} {input.trim1} {input.trim2} &>> {log}
echo "Renaming results from trimmed fastq analysis" >> {log} # Renames files because we can't choose fastqc output
mv {params.html1_after} {output.html1_after} 2>> {log}
mv {params.zipfile1_after} {output.zipfile1_after} 2>> {log}
mv {params.html2_after} {output.html2_after} 2>> {log}
mv {params.zipfile2_after} {output.zipfile2_after} 2>> {log}
echo "Results saved in <results/{wildcards.sample}/fastqc_reports/>" >> {log}
'''
This solution is very long and much more complicated than the other one. However, it makes up for the complexity by allowing a total control on what is happening: with this method, we can choose where the temporary files are saved and the names of the outputs. It could have been shortened by using -o . to tell FastQC to create the files in the current working directory instead of a specific one, but this would have created another problem: if we run multiple jobs in parallel, then Snakemake may potentially try to produce files from different jobs but with the same temporary destination. In this case, the different instances would be trying to write to the same temporary files at the same time, overwriting each other and corrupting the output files.
Several interesting things are happening in both versions of this rule:
- Much like for the outputs, it is possible to refer to the inputs of a rule directly in another rule with the syntax
rules.<rule_name>.input.<input_name> - FastQC doesn’t create the output directory by itself (other programs might insist that the output directory does not already exist), so we have to create it manually with
mkdirin the shell command before running FastQC - The
-pflag ofmkdirmake parent directories as needed and does not return an error if the directory already exists
Directory creation
Remember that in most cases it is not necessary to manually create directories because Snakemake will do it for you. Even when using a directory() output, Snakemake will not create the directory itself but most applications will make the directory for you; FastQC is an exception.
Hint
If you want to make sure that a certain rule is executed before another, you can write the outputs of the first rule as inputs of the second one, even if you don’t use them in the rule. For example, we could force the execution of FastQC before mapping the reads with only a few modifications to rule read_mapping:
rule read_mapping:
'''
This rule maps trimmed reads of a fastq on a reference assembly.
'''
input:
trim1 = rules.fastq_trim.output.trim1,
trim2 = rules.fastq_trim.output.trim2, # Do not forget to add a comma here
fastqc = rules.fastq_qc_sol4.output.html1_before # This single line will force the execution of FASTQC before read mapping
output:
sam = 'results/{sample}/{sample}_mapped_reads.sam',
report = 'results/{sample}/{sample}_mapping_report.txt'
params:
index = 'resources/genome_indices/Scerevisiae_index'
log:
'logs/{sample}/{sample}_mapping.log'
benchmark:
'benchmarks/{sample}/{sample}_mapping.txt'
resources:
mem_gb = 2
threads: 4
shell:
'''
echo "Mapping the reads" > {log}
hisat2 --dta --fr --no-mixed --no-discordant --time --new-summary --no-unal \
-x {params.index} --threads {threads} \
-1 {input.trim1} -2 {input.trim2} -S {output.sam} --summary-file {output.report} 2>> {log}
echo "Mapped reads saved in <{output.sam}>" >> {log}
echo "Mapping report saved in <{output.report}>" >> {log}
'''
Modularising a workflow
If you keep developing a workflow long enough, you are bound to encounter some cluttering problems. Have a look at your current Snakefile: with only 5 rules, it is already almost 200 lines long. Imagine what happens when your workflow comprises dozens of rules?! The Snakefile may become messy and harder to maintain and edit. This is why it quickly becomes crucial to modularise your workflow; this is a common practice in programming in general. This approach also makes it easier to re-use pieces of workflow in the future. Modularisation comes at 4 different levels:
- The most fine-grained level are wrappers. Wrappers allow to quickly use popular tools and libraries in Snakemake workflows, thanks to the
wrapperdirective. Wrappers are automatically downloaded and deploy a conda environment when running the workflow, which increases reproducibility, however their implementation can sometimes be ‘rigid’ and you may have to write your own rule. See the official documentation for more explanations - For larger, reusable parts belonging to the same workflow, it is recommended to write smaller snakefiles and include them into a main Snakefile with the
includestatement. Note that in this case, all rules share a common config file. See the official documentation for more explanations - The next level of modularisation is provided via the
modulestatement, which enables arbitrary combination and re-use of rules in the same workflow and between workflows. See the official documentation for more explanations - Finally, Snakemake also provides a syntax to define subworkflows, but this syntax is currently being deprecated in favor of the
modulestatement. See the official documentation for more explanations
In this course, we will only use the 2nd level of modularisation. In more details, the idea is to write a main Snakefile in workflow/Snakefile, to place the other snakefiles containing the rules in the subfolder workflow/rules (these ‘sub-Snakefile’ should end with .smk, the recommended file extension of Snakemake) and to tell Snakemake to import the modular snakefiles in the main Snakefile with the include: <path/to/snakefile.smk> syntax.
Rules organisation
How to organize rules is up to you, but a common approach would be to create “thematic” modules, i.e. regroup rules involved in the same general step of the workflow.
Exercise: Move your current Snakefile into the subfolder workflow/rules and rename it to read_mapping.smk. Then create a new Snakefile in workflow/ and import read_mapping.smk in it using the include syntax. You should also move the importation of the config file from the modular Snakefile to the main one.
Answer
We will solve this problem step by step. First, create the new file structure:
mkdir workflow/rules # Create a new folder
mv workflow/Snakefile workflow/rules/read_mapping.smk # Move and rename the modular snakefile
touch workflow/Snakefile # Recreate the main Snakefile
Then, fill the main Snakefile with include and configfile:
'''
Main Snakefile of the RNAseq analysis workflow. This workflow can clean and
map reads, and perform Differential Expression Analyses.
'''
# Path of the config file
configfile: 'config/config.yaml'
# Rules to execute the workflow
include: 'rules/read_mapping.smk'
Finally, do not forget to remove the config file import (configfile: 'config/config.yaml') from the snakefiles (workflow/rules/read_mapping.smk)
Relative paths
- Includes are relative to the directory of the Snakefile in which they occur. For example, if the Snakefile resides in
workflow, then Snakemake will search for the included snakefiles inworkflow/path/to/other/snakefile, regardless of the working directory - You can place snakefiles in a sub-directory without changing input and output paths, as these paths are relative to the working directory. However, you will need to edit paths to external scripts and conda environments, as these paths are relative to the snakefile from which they are called (this will be discussed in the last series of exercises)
In practice, you can imagine that the line include: <path/to/snakefile.smk> is replaced by the entire content of snakefile.smk in Snakefile. This means that syntaxes like rules.<rule_name>.output.<output_name> can still be used in snakefiles, even if the rule <rule_name> was defined in another snakefile, as long as the snakefile in which <rule_name> is defined is included before the snakefile that uses rules.<rule_name>.output. This also works for input and output functions.
Using a target rule and aggregating outputs
Creating a target rule
Modularisation also offers a great opportunity to facilitate the execution of the workflow. By default, if no target is given at the command line, Snakemake executes the first rule in the Snakefile. Hence, we have always executed the workflow by specifying a target file in the command line to avoid this behaviour. But we can actually use this property to make the execution easier by writing a pseudo-rule (also called target-rule and usually named rule all) in the Snakefile which has all the desired outputs (or a particular subsets of them) files as input files. This rule will look like this:
rule all:
input:
'path/to/ouput1',
'path/to/ouput2'
Order of rules in Snakefile/snakefiles
Apart from Snakemake considering the first rule of the workflow as the default target, the order of rules in the Snakefile/snakefiles is arbitrary and does not influence the DAG of jobs.
Exercise: Implement a special rule in the Snakefile so that the final output is generated by default when running snakemake without specifying a target, then test your workflow with a dry-run.
Hint
- Remember that a rule is not required to have an output nor a shell command
- The inputs of
rule allshould be the final outputs that you want to generate (those from the last rule you wrote)
Answer
If we consider that the last outputs are the ones produced by rule reads_quantification_genes, we can write the target rule like this:
# Master rule that launches the workflow
rule all:
'''
Dummy rule to automatically generate the required outputs.
'''
input:
'results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv'
Note that we used only one of the two outputs of rule reads_quantification_genes. We do this because it is enough to trigger the execution and if the rule didn’t produce both outputs, Snakemake would crash and report it this error.
Now, let’s try to do a dry-run with this new rule: snakemake --cores 4 -F -r -p -n. You should see all the rules appearing thanks to the -F flag, including:
localrule all:
input: results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv
jobid: 0
reason: Input files updated by another job: results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv
resources: tmpdir=/tmp
Job stats:
job count
-------------------------- -------
all 1
fastq_qc_sol4 1
fastq_trim 1
read_mapping 1
reads_quantification_genes 1
sam_to_bam 1
total 6
Aggregating outputs
Using a target rule like the one presented in the previous paragraph gives another opportunity to make things easier. In the rule we just created, we used a hard-coded input and by now, you should know that this is not an optimal solution and that we should avoid this as much as possible, especially if you have many samples to process. To solve this problem, we will rely on the expand function.
Exercise: Write an expand() syntax to generate a list of outputs from rule reads_quantification_genes with all the RNAseq samples. What do you need to write this?
Answer
The output of rule reads_quantification_genes has the following syntax: 'results/{sample}/{sample}_genes_read_quantification.tsv'.
First, we need to create a Python list containing all the values that the {sample} wildcards can take:
SAMPLES = ['highCO2_sample1', 'highCO2_sample2', 'highCO2_sample3', 'lowCO2_sample1', 'lowCO2_sample2', 'lowCO2_sample3']
Then, we can transform the output syntax with expand():
expand('results/{sample}/{sample}_genes_read_quantification.tsv', sample=SAMPLES)
Exercise: Use these two elements (the list of samples and the expand() syntax) in the target rule to ask Snakemake to generate all the outputs.
Answer
You need to add the sample list to the Snakefile before the rule all and replace the value of the input directive:
# Sample list
SAMPLES = ['highCO2_sample1', 'highCO2_sample2', 'highCO2_sample3', 'lowCO2_sample1', 'lowCO2_sample2', 'lowCO2_sample3']
# Master rule that launches the workflow
rule all:
'''
Dummy rule to automatically generate the required outputs.
'''
input:
expand('results/{sample}/{sample}_genes_read_quantification.tsv', sample=SAMPLES)
If you launch the workflow in dry-run mode with this new rule: snakemake --cores 4 -F -r -p -n. You should see all the rules appearing 5 times (1 for each sample that hasn’t been processed yet):
localrule all:
input: results/highCO2_sample1/highCO2_sample1_genes_read_quantification.tsv, results/highCO2_sample2/highCO2_sample2_genes_read_quantification.tsv, results/highCO2_sample3/highCO2_sample3_genes_read_quantification.tsv, results/lowCO2_sample1/lowCO2_sample1_genes_read_quantification.tsv, results/lowCO2_sample2/lowCO2_sample2_genes_read_quantification.tsv, results/lowCO2_sample3/lowCO2_sample3_genes_read_quantification.tsv
jobid: 0
reason: Input files updated by another job: results/lowCO2_sample1/lowCO2_sample1_genes_read_quantification.tsv, results/lowCO2_sample2/lowCO2_sample2_genes_read_quantification.tsv, results/lowCO2_sample3/lowCO2_sample3_genes_read_quantification.tsv, results/highCO2_sample3/highCO2_sample3_genes_read_quantification.tsv, results/highCO2_sample2/highCO2_sample2_genes_read_quantification.tsv
resources: tmpdir=/tmp
Job stats:
job count
-------------------------- -------
all 1
fastq_qc_sol4 5
fastq_trim 5
read_mapping 5
reads_quantification_genes 5
sam_to_bam 5
total 26
But we can do even better! At the moment, samples are defined in a list at the top of the Snakefile. To further improve the workflow’s usability, we can define samples in the config file, so they can easily be added, removed, or modified by the user.
Exercise: Implement a parameter in the config file to specify sample names and modify rule all to use this parameter in the expand() syntax.
Answer
First, we need to modify the config file:
# Configuration options of RNAseq-analysis workflow
# Location of the genome indices
index: 'resources/genome_indices/Scerevisiae_index'
# Location of the annotation file
annotations: 'resources/Scerevisiae.gtf'
# Sample names
samples:
- highCO2_sample1
- highCO2_sample2
- highCO2_sample3
- lowCO2_sample1
- lowCO2_sample2
- lowCO2_sample3
Then, we need to use the config file in the expand() syntax (and remove SAMPLES from the Snakefile, because we don’t need this variable anymore):
# Master rule that launches the workflow
rule all:
'''
Dummy rule to automatically generate the required outputs.
'''
input:
expand('results/{sample}/{sample}_genes_read_quantification.tsv', sample=config['samples'])
Here, config['samples'] is a Python list containing strings, each string being a sample name. This is because a list of parameters become a list during the config file parsing.
An even more Snakemake-idiomatic solution
There is an even better and more Snakemake-idiomatic version of the expand() syntax:
expand(rules.reads_quantification_genes.output.gene_level, sample=config['samples']).
While it may not seem easy to use and understand, this entirely removes the need to write the output paths!
Running the other samples of the workflow
Exercise: Touch the files already present in your workflow to avoid re-creating them and then run your workflow on the 5 other samples.
Answer
- Touch the existing files:
snakemake --cores 1 --touch - Run the workflow
snakemake --cores 4 -r -p
Thanks to the parallelisation, the workflow execution should take less than 10 min in total to process all the samples!
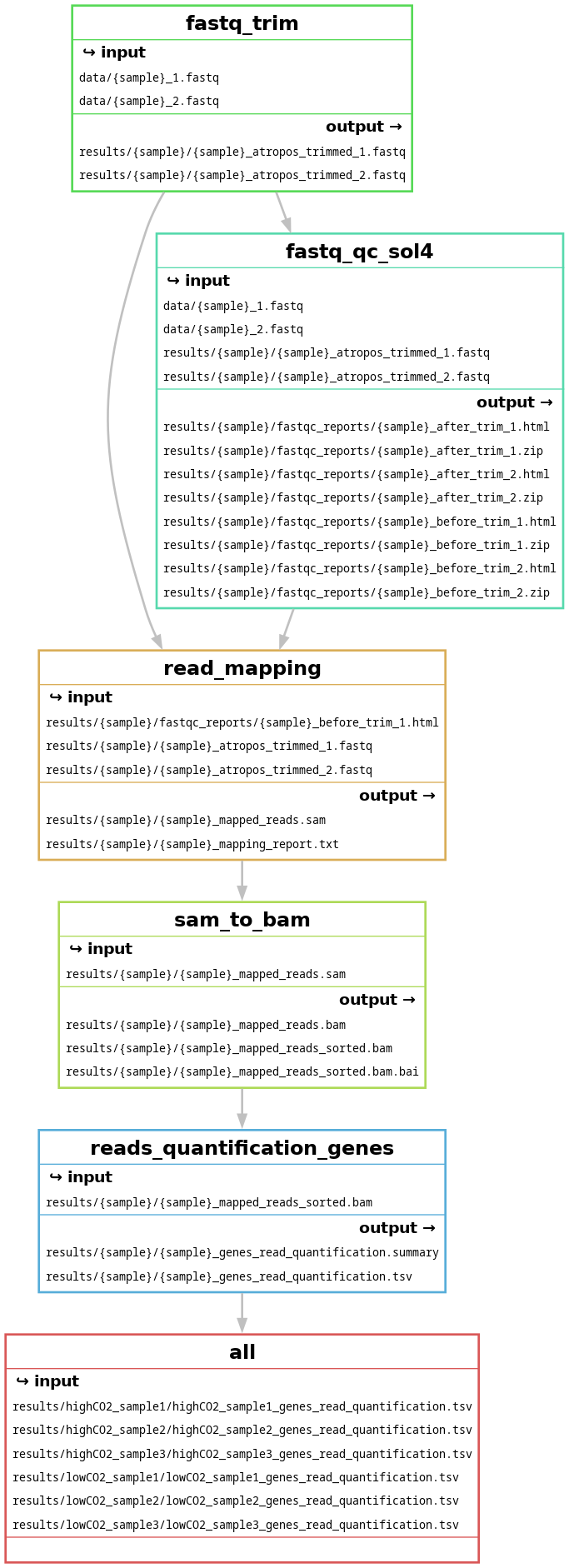
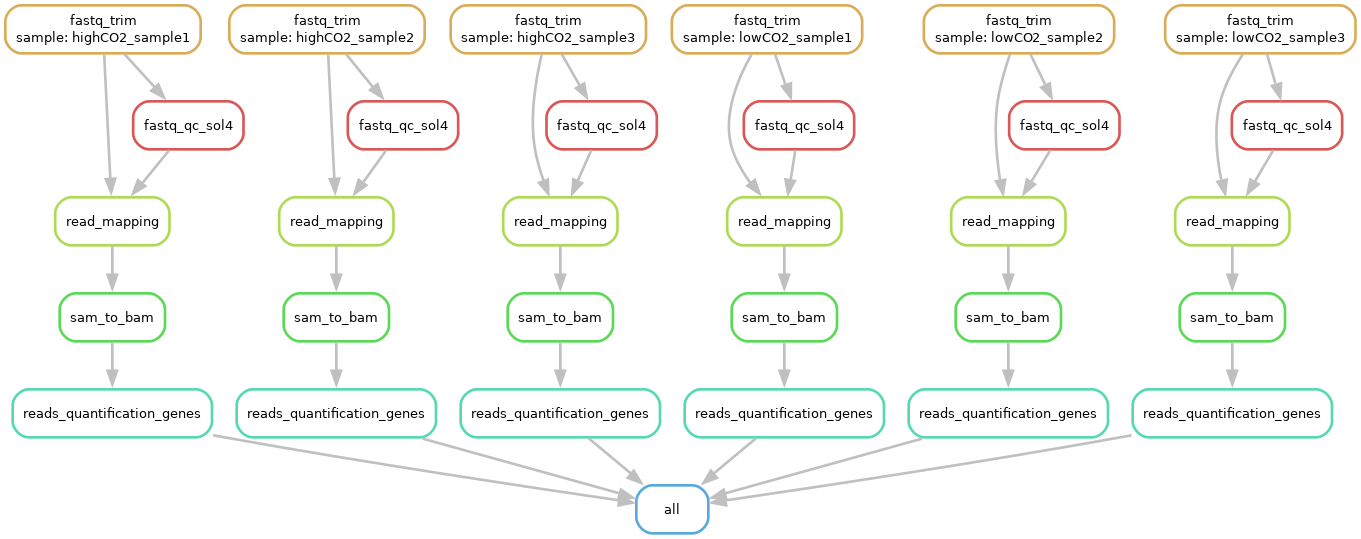
Exercise: Generate the workflow DAG and filegraph.
Answer
- Generate the DAG:
snakemake --cores 1 -F -r -p --rulegraph | dot -Tpng > images/all_samples_rulegraph.png - Generate the filegraph:
snakemake --cores 1 -F -r -p --filegraph | dot -Tpng > images/all_samples_filegraph.png
Your DAG should resemble this:
And this should be your filegraph (open the picture in a new tab to zoom in):