Being reproducible with Snakemake
Learning outcomes
After having completed this chapter you will be able to:
- Create and use an input function
- Deploy a conda environment and run scripts within it
- Deploy a Docker/Singularity container and run scripts within it
Material
Reproducibility in Snakemake:
Additional advanced concepts:
Exercises
In this series of exercises, we will create the last two rules of the workflow. Each rule will execute a script, one in Python and one in R, and both rules will have dedicated environment that you will need to take into account in the snakefiles. These last two rules are quite different from the previous ones, so it would be a good idea to implement them in a new snakefile in workflow/rules, for example called analysis.smk.
Development and back-up
During this session, we will modify our snakefiles quite heavily, so it may be a good idea to start by making a back-up: cp -r worklow/ worklow_backup. As a general rule, if you have a doubt on the code you are developing, do not hesitate to make a back-up.
Hint
This is not a programming course, so you won’t need to write the scripts: they were already prepared for you!
Creating a rule to gather read count files
To perform a Differential Expression Analysis (DEA), it is easier to have a single file gathering all the read counts of the different samples.
Exercise: Implement a rule to list and merge read count files (coming from rule reads_quantification_genes) into a single file using the Python script provided [here]https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/scripts/solutions/day2/session4/workflow/scripts/count_table.py).
Information on the script to compute the table
- You can download the script with
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/scripts/solutions/day2/session4/workflow/scripts/count_table.py - It goes in a special directory in your workflow
- It is written in Python
- It takes a list of files as input, each file being the read count output of featureCounts
- It produces one output, a tab-separated table containing all the read counts of the different sample gathered by gene
Hint
While the goal of this rule is quite easy to grasp, setting it up requires using several advanced notions of Snakemake, so here is a little outline of the steps you should take:
- Build the basic structure of your rule: name, output, log, benchmark
- Memory should be set at 500 MB
- Threads should be set at 1
- Don’t focus on the inputs for now
- Think about the directive you want to use to run the script
- Looking at the script length with
wc -l <path/to/script>could help you decide
- Looking at the script length with
- Think about the location/path of the script
- Check the beginning of the script to see if you need any special Python packages. You can do that with
head <path/to/script>. If you see lines containingimport <package_name>, it means that the script is using external Python packages- If the script is using external packages, think on how can you provide them
- Finally, identify the inputs your rule needs
- Think on how you can easily list and provide the inputs. Does it remind you of something you saw in the course?
Now, let’s solve these problems one by one!
Building the rule structure
You have done that a few times already, so it should not be too difficult.
Exercise: Set the output, log, benchmark, resources and thread values.
Answer
rule count_table:
'''
This rule merges all the gene count tables of an assembly into one table.
'''
input:
?
output:
count_table = 'results/total_count_table.tsv'
log:
'logs/total_count_table.log'
benchmark:
'benchmarks/total_count_table.txt'
resources:
mem_mb = 500
threads: 1
?:
?
Note that we do not need to use wildcards in the output name: only one file will be created, and its name will not change depending on the sample name, because we use all the samples to create it.
Getting the Python script and running it
Exercise: Download the script and place it the proper folder. Remember that per the official documentation, scripts should be stored in a subfolder workflow/scripts.
Answer
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/scripts/solutions/day2/session4/workflow/scripts/count_table.py # Download the script
mkdir -p workflow/scripts # Create the appropriate folder
mv count_table.py workflow/scripts # Move the script in the newly created folder
Exercise: Check how long the script is.
Answer
If you run wc -l workflow/scripts/count_table.py, you will see that the script is 67 lines long. This is too much to use a run directive, so we will use the script directive instead. This means that we need to add the following to our rule count_table:
script:
'../scripts/count_table.py'
Script path
If you placed included files in subfolders (like rules/analysis.smk), you need to change relative paths for external script files, hence the ../ in the script path.
Hint
In many cases, it would be nice to have a script that can be called by Snakemake but also work with standard Python, so that the code can be reused in other projects. There are several ways to do that:
- You could implement most of the functionalities in a module and use this module in a simple script called by Snakemake
- You could test for the existence of a
snakemakeobject and handle parameter values differently (e.g. command-line arguments) if the object does not exist
Inside the script, you have access to an object snakemake that provides access to the same objects that are available in the run and shell directives (input, output, params, wildcards, log, threads, resources, config), e.g. you can use snakemake.input[0] to access the first input file of a rule, or snakemake.input.input_name to access a specific named input.
Exercise: Check the script content to see whether it requires specific packages.
Answer
If you run head workflow/scripts/count_table.py, you will see several import commands at the start of the script, including import pandas as pd. pandas is a great package, but it is not part of the default packages natively shipped with Python. This means that we need to find a solution to provide it to the rule. The easiest way to do that is to create a conda environment dedicated to the rule. Conda environments should be stored in a subfolder workflow/envs.
Create the appropriate folder: mkdir -p workflow/envs. Then, write the following configuration in the environment file, workflow/envs/py.yaml:
# Environment file to perform data processing with python
name : python
channels :
- conda-forge
- bioconda
dependencies :
- python >= 3.10
- pandas == 1.4.3
This means that you need to add the following to your rule count_table:
conda:
'../envs/py.yaml'
Environment file path
If you placed included files in subfolders (like rules/analysis.smk), you need to change relative paths for conda environments files as well, hence the ../ in the environment file path.
Using conda environments improves reproducibility for many reasons, including version control and the fact that users do not need to manually manage software dependencies. Note: the first execution of the workflow after adding Conda environments will take some time, because Conda will have to download and install all the software.
Identifying and listing the input files
The before-last step is the most complex one: identifying all the inputs of the rule and gathering them in a list. Here, there are only 6 samples, so in theory, we could list them directly… However, by now you should that is not a good solution. Fortunately, there is a much more elegant and convenient way to do this: an input function, which provides the added benefit of scaling up very well if the number of samples increase.
We already wrote the input function for you:
# Input function used in rule count_table
def get_gene_counts(wildcards):
'''
This function lists all the gene count tables of samples in the config file
'''
# Note that here {sample} is not a wildcard, it is an f-string variable!
return [f"results/{sample}/{sample}_genes_read_quantification.tsv"
for sample in config['samples']]
This function will loop over the list of samples in the config file, replace ‘{sample}’ with the current sample name of the iteration to create a string which is the output path from the rule reads_quantification_genes of said sample. Then, it will aggregate all the paths in a list and return this list.
Details on input functions
- Input functions take the
wildcardsglobal object as single argument - You can access wildcard values inside an input function with the syntax
{wildcards.wildcards_name} - Input and output functions can return a list of files, which will then be automatically handled like multiple inputs or outputs by Snakemake. These functions can also return a dictionary; in this case, the function should be called with the syntax
input: unpack(<function_name>). The dictionary’s keys will be interpreted as input/output names and the dictionary’s values will be interpreted as input/output values - Functions are evaluated before the workflow is executed. As a consequence, they cannot be used to list the content of an output directory, since the directory does not exist before the workflow is executed!
Exercise: Insert the function in the proper Snakefile and adapt the input value of the rule accordingly.
Answer
There are two things to do:
* Insert the input function in workflow/rules/analysis.smk, before the rule, otherwise you will get a name 'get_gene_counts' is not defined error (the function needs to be defined before Snakemake looks for it when it parses the rule input).
* Use the function name as value for the input directive
Your rule and function should resemble this:
# Input function used in rule count_table
def get_gene_counts(wildcards):
'''
This function lists all the gene count tables of samples in the config file
'''
return [f"results/{sample}/{sample}_genes_read_quantification.tsv"
for sample in config['samples']]
rule count_table:
'''
This rule merges all the gene count tables of an assembly into one table.
'''
input:
get_gene_counts
output:
count_table = 'results/total_count_table.tsv'
log:
'logs/total_count_table.log'
benchmark:
'benchmarks/total_count_table.txt'
conda:
'../envs/py.yaml'
resources:
mem_mb = 500
threads: 1
script:
'../scripts/count_table.py'
Input functions usage
You don’t need to use parentheses or specify any argument when you call an input function in the input directive.
Adapting the Snakefile and running the rule
Now, all that is left is to run the rule to create the table.
Exercise: Which command should you use to create the output? Is there anything else to do beforehand?
Answer
It turns out that we cannnot launch the workflow directly: we need to include the new rule file in the Snakefile and adapt the output of the rule all! Your Snakefile should now resemble this:
'''
Main Snakefile of the RNAseq analysis workflow. This workflow can clean and
map reads, and perform Differential Expression Analyses.
'''
# Path of the config file
configfile: 'config/config.yaml'
# Rules to execute the workflow
include: 'rules/read_mapping.smk'
include: 'rules/analyses.smk'
# Master rule that launches the workflow
rule all:
'''
Dummy rule to automatically generate the required outputs.
'''
input:
'results/total_count_table.tsv'
Finally, run the workflow with snakemake --cores 4 -r -p --use-conda
–use-conda
Do not forget --use-conda otherwise Snakemake will not use the environments!!!
Creating a rule to detect Differentially Expressed Genes
It is now time to write the final rule of the workflow. This rule will perform the DEA using the global count table we previously created.
Exercise: Implement a rule to perform DEA using the R script provided here.
Information on the script to compute the table
- You can download the script with
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/scripts/solutions/day2/session4/workflow/scripts/DESeq2.R - It goes in a special directory in your workflow
- It is written in R
- It takes a global read counts table as input
- It produces two outputs:
- A tab-separated table containing the DEG and the associated statistical results
- A pdf file containing control plots of the analysis
Hint
While not being trivial, this rule is much easier than the previous one and some things work similarly. Still, here is a little outline of the steps you should take:
- Build the basic structure of your rule: name, input, outputs, log, benchmark
- Memory should be set at 1 GB
- Threads should be set at 2
- Think about the directive you want to use to run the script
- Remember than there is only one way to run an R script
- Think about the location/path of the script
- This script is using a lot of external packages. Fortunately, all these packages are available in a certain Docker image you worked with yesterday
Now, let’s write this last rule!
Building the rule structure
You have done that a few times already, so it should not be too difficult.
Exercise: Set the input, outputs, log, benchmark, resources and thread values.
Answer
rule differential_expression:
'''
This rule detects DEGs and plots associated visual control graphs (PCA,
heatmaps...).
'''
input:
table = rules.count_table.output.table
output:
deg = 'results/deg_list.tsv',
pdf = 'results/deg_plots.pdf'
log:
'logs/differential_expression.log'
benchmark:
'benchmarks/differential_expression.txt'
?:
?
resources:
mem_gb = 1
threads: 2
?:
?
Note that we do not need to use wildcards in this rule, because all the files are precisely defined.
Getting the R script and running it
Exercise: Download the script and place it the proper folder. Remember that per the official documentation, scripts should be stored in a subfolder workflow/scripts.
Answer
wget https://raw.githubusercontent.com/sib-swiss/containers-snakemake-training/main/scripts/solutions/day2/session4/workflow/scripts/DESeq2.R # Download the script
mv DESeq2.R workflow/scripts # Move the script in the newly created folder
Exercise: Find a way to run the script.
Answer
There is only one way to run an R script: use the script directive. This means that we need to add the following to our rule differential_expression:
script:
'../scripts/DESeq2.R'
Script path
If you placed included files in subfolders (like rules/analysis.smk), you need to change relative paths for external script files, hence the ../ in the script path.
Hint
Inside the script, an S4 object named snakemake analogous to the Python case available and allows access to input and output files and other parameters. Here the syntax follows that of S4 classes with attributes that are R lists, e.g. you can access the first input file with snakemake@input[[1]] (note that the first file does not have index 0 here, because R starts counting from 1). Named input and output files can be accessed in the same way, by just providing the name instead of an index, e.g. snakemake@input[["myfile"]].
Exercise: Find an efficient way to create a computing environment for the rule.
Hint
Remember what you did during Day 1, session 3 “Working with Dockerfiles”!
Answer
During Day 1, you built your own docker image, called deseq2. This image actually contains everything we need to run DEA, so let’s use it again, but with Snakemake this time! This means that you need to add the following to your rule differential_expression:
container:
'docker://geertvangeest/deseq2:v1'
Your own Docker image
First try with your own image. If it doesn’t work, then you can use Geert’s image: geertvangeest/deseq2:v1.
After all these modifcations, this is what your final rule should look like:
Answer
rule differential_expression:
'''
This rule detects DEGs and plots associated visual control graphs (PCA,
heatmaps...).
'''
input:
table = rules.count_table.output.table
output:
deg = 'results/deg_list.tsv',
pdf = 'results/deg_plots.pdf'
log:
'logs/differential_expression.log'
benchmark:
'benchmarks/differential_expression.txt'
container:
'docker://geertvangeest/deseq2:v1'
resources:
mem_gb = 1
threads: 2
script:
'../scripts/DESeq2.R'
Adapting the Snakefile and running the rule
Now, all that is left is to run the rule to create the DEG list.
Exercise: Which command should you use to create the output? Is there anything else to do beforehand?
Answer
It turns out that we cannnot launch the workflow directly: we need to include the new rule file in the Snakefile and adapt the output of the rule all! Your Snakefile should now resemble this:
'''
Main Snakefile of the RNAseq analysis workflow. This workflow can clean and
map reads, and perform Differential Expression Analyses.
'''
# Path of the config file
configfile: 'config/config.yaml'
# Rules to execute the workflow
include: 'rules/read_mapping.smk'
include: 'rules/analyses.smk'
# Master rule that launches the workflow
rule all:
'''
Dummy rule to automatically generate the required outputs.
'''
input:
'results/deg_list.tsv'
Finally, run the workflow with snakemake --cores 4 -r -p --use-singularity. You should see new Snakemake information messages:
Rscript --vanilla /path/to/snakemake_rnaseq/.snakemake/scripts/tmpge97d_lz.DESeq2.R
Activating singularity image /path/to/snakemake_rnaseq/.snakemake/singularity/8bfdbe93244feb95887ab5d33a705017.simg
INFO: squashfuse not found, will not be able to mount SIF
INFO: fuse2fs not found, will not be able to mount EXT3 filesystems
INFO: gocryptfs not found, will not be able to use gocryptfs
INFO: Converting SIF file to temporary sandbox...
INFO: Cleaning up image...
–use-singularity
Do not forget --use-singularity otherwise Snakemake will not pull the image!!!
Hint
If you want to see how a Snakemake-generated Dockerfile looks like, use: snakemake --cores 1 --containerize > Dockerfile
Exercise: How many DEG were detected?
Answer
Have a look at the list that was just created: cat results/deg_list.tsv. 8 genes are differentially expressed!
Exercise: If you had to re-run the entire workflow from scratch, what command would you use?
Answer
You would need to execute snakemake --cores 4 -r -p --use-conda --use-singularity -F.
* -F is to force the execution of the entire workflow
* Don’t forget --use-conda --use-singularity! Otherwise, you will lack some software and packages and the workflow will crash!
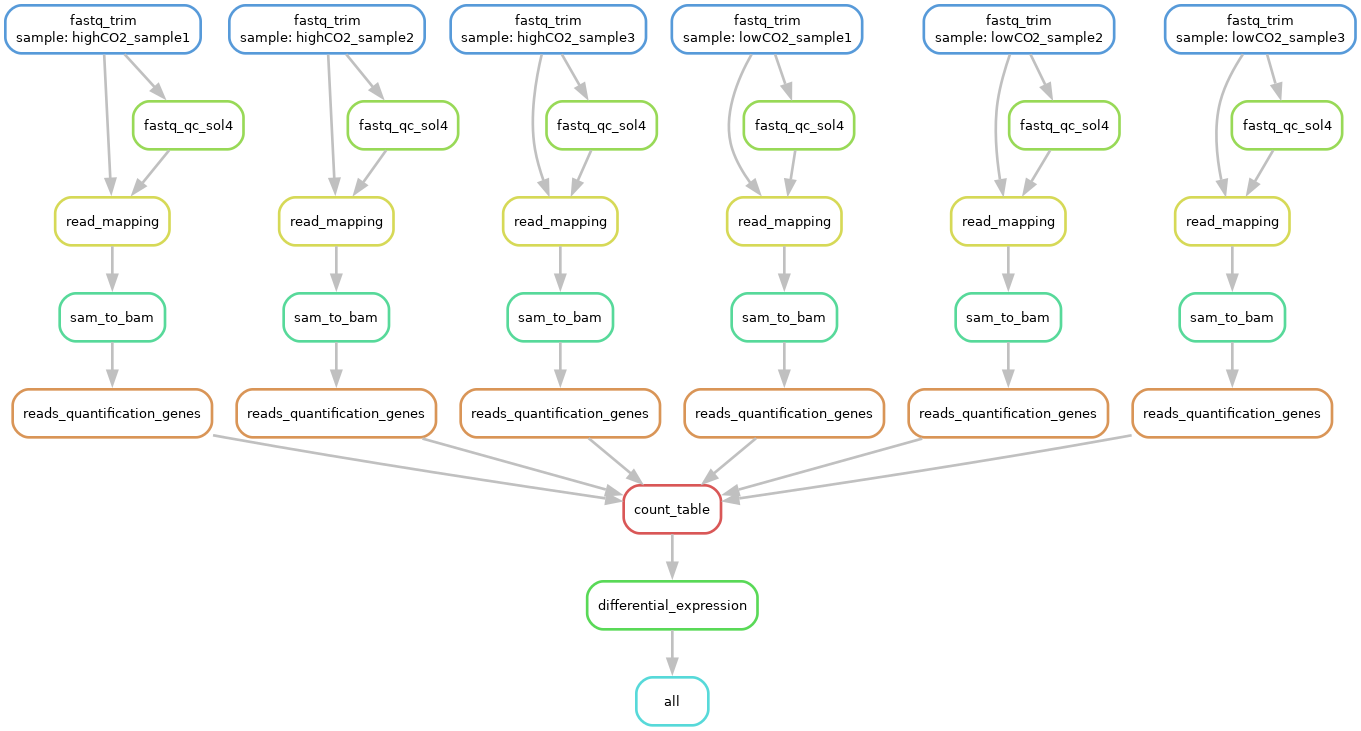
Exercise: Visualise the DAG of the entire workflow.
Answer
You should now be used to this. snakemake --cores 1 -r -p -F --dag | dot -T png > images/total_dag.png
This is the DAG you should see:
Congratulations, you are now able to create a Snakemake workflow and make it reproducible thanks to conda/mamba and Docker/Singularity! To make things even better, have a look at Snakemake’s best practices!