Integration
Material
Exercises
Let’s have a look at the UMAP again. Although cells of different samples are shared amongst ‘clusters’, you can still see seperation within the clusters:
Seurat::DimPlot(seu, reduction = "umap")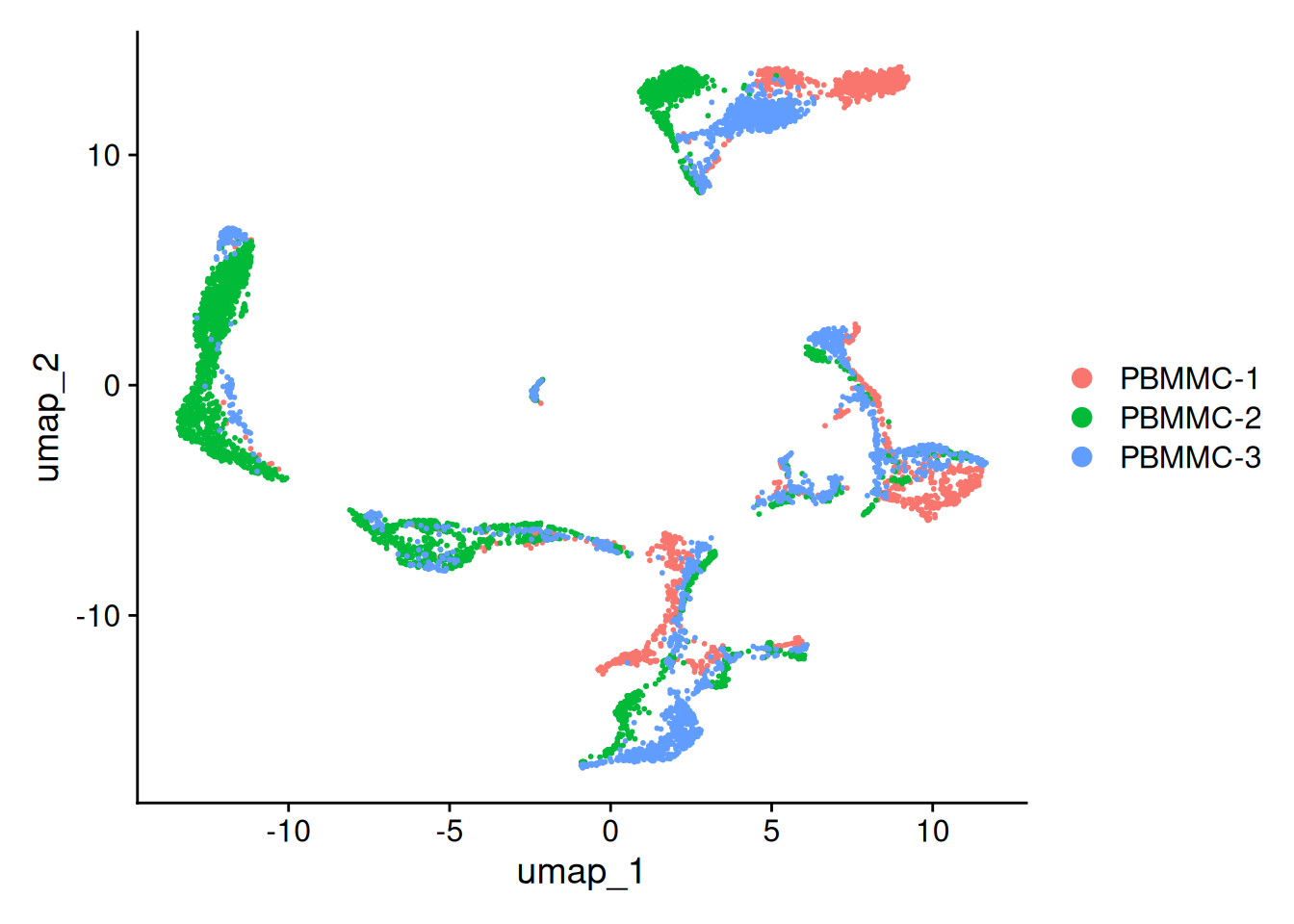
Let’s also make the function again for 3D UMAP
library(plotly)
seu <- Seurat::RunUMAP(seu, dims = 1:25, n.components = 3, reduction.name = "umap_3D")
plot_3d_umap <- function(seu,
reduction = "umap_3D",
sample_col = "orig.ident",
point_size = 4,
point_opacity = 0.5) {
## 1. Extract coordinates -------------------------------------------------
umap_coords <- as.data.frame(Embeddings(seu, reduction = reduction))
umap_coords$Sample <- seu@meta.data[[sample_col]]
umap_coords$Cell <- rownames(umap_coords) # for hover
## 2. Build Plotly object --------------------------------------------------
p <- plot_ly(
data = umap_coords,
x = ~umap3D_1, y = ~umap3D_2, z = ~umap3D_3,
color = ~Sample, # default Plotly palette
type = "scatter3d",
mode = "markers",
marker = list(
size = point_size,
opacity = point_opacity
),
text = ~paste("Cell:", Cell, "<br>Sample:", Sample),
hovertemplate = "<b>%{text}</b><extra></extra>"
) %>%
layout(
title = list(
text = "<b>3D UMAP of Cells by Sample</b>",
font = list(size = 18, family = "Arial, sans-serif"),
x = 0.5, xanchor = "center", y = 0.95
),
scene = list(
xaxis = list(
title = "<b>UMAP 1</b>", titlefont = list(size = 14),
showgrid = TRUE, gridcolor = "rgba(200,200,200,0.5)",
backgroundcolor = "rgba(245,245,255,0.9)",
zerolinecolor = "rgba(100,100,100,0.3)"
),
yaxis = list(
title = "<b>UMAP 2</b>", titlefont = list(size = 14),
showgrid = TRUE, gridcolor = "rgba(200,200,200,0.5)",
backgroundcolor = "rgba(245,245,255,0.9)",
zerolinecolor = "rgba(100,100,100,0.3)"
),
zaxis = list(
title = "<b>UMAP 3</b>", titlefont = list(size = 14),
showgrid = TRUE, gridcolor = "rgba(200,200,200,0.5)",
backgroundcolor = "rgba(245,245,255,0.9)",
zerolinecolor = "rgba(100,100,100,0.3)"
),
camera = list(eye = list(x = 1.5, y = 1.5, z = 1.5)),
aspectmode = "cube"
),
legend = list(
title = list(text = "<b>Sample</b>", font = list(size = 12)),
bgcolor = "rgba(255,255,255,0.8)",
bordercolor = "gray", borderwidth = 1
),
margin = list(l = 0, r = 0, t = 60, b = 0),
paper_bgcolor = "rgba(250,250,252,1)",
plot_bgcolor = "rgba(250,250,252,1)"
) %>%
config(
displayModeBar = TRUE,
displaylogo = FALSE,
modeBarButtonsToRemove = c("sendDataToCloud", "lasso2d", "select2d")
)
return(p)
}
plot_3d_umap(seu = seu, reduction = "umap_3D", sample_col = "orig.ident")To perform the integration, we split our object by sample, resulting into a set of layers within the RNA assay. The layers are integrated and stored in the reduction slot - in our case we call it integrated.cca. Then, we re-join the layers
seu[["RNA"]] <- split(seu[["RNA"]], f = seu$orig.ident)
seu <- Seurat::IntegrateLayers(object = seu, method = CCAIntegration,
orig.reduction = "pca",
new.reduction = "integrated.cca",
verbose = FALSE)
# re-join layers after integration
seu[["RNA"]] <- JoinLayers(seu[["RNA"]])# 1. Ensure default assay & have your HVGs / PCA ready
DefaultAssay(seu) <- "SCT"
# If pcaSCT is missing or outdated:
seu <- RunPCA(seu, reduction.name = "pcaSCT", verbose = FALSE)Warning: Key 'PC_' taken, using 'pcasct_' instead# 2. Create a new v5 assay from your current SCT (copies data/counts/scale.data)
# → SCT residuals go into 'data' layer; this becomes Assay5 class
seu[["SCT_v5"]] <- as(object = seu[["SCT"]], Class = "Assay5")Warning: Key 'sct_' taken, using 'sctv5_' instead# Confirm class & layers
class(seu[["SCT_v5"]]) # should be "Assay5"[1] "Assay5"
attr(,"package")
[1] "SeuratObject"Layers(seu[["SCT_v5"]]) # expect "counts", "data", possibly "scale.data"[1] "data" "counts" "scale.data"# Optional: If you want split layers (recommended for integration):
seu[["SCT_v5"]] <- split(seu[["SCT_v5"]], f = seu$orig.ident)Splitting 'counts', 'data' layers. Not splitting 'scale.data'. If you would like to split other layers, set in `layers` argument.Layers(seu[["SCT_v5"]]) # now per-batch layers (counts.PBMMC-1, data.PBMMC-1, etc.)[1] "data.PBMMC-1" "data.PBMMC-2" "data.PBMMC-3" "scale.data"
[5] "counts.PBMMC-1" "counts.PBMMC-2" "counts.PBMMC-3"# 3. Run CCAIntegration on the v5 assay
seu <- IntegrateLayers(
object = seu,
assay = "SCT_v5", # ← use the new v5 assay here
method = CCAIntegration,
orig.reduction = "pcaSCT", # your existing PCA (computed on SCT)
new.reduction = "SCT.cca",
features = VariableFeatures(seu), # still good to restrict
verbose = FALSE
)Warning: Adding a dimensional reduction (SCT.cca) without the associated assay
being presentDefaultAssay(seu) <- "RNA"We can then use this new integrated matrix for clustering and visualization. Now, we can re-run and visualize the results with UMAP.
Create the UMAP again on the integrated.cca reduction (using the function RunUMAP - set the option reduction accordingly). After that, generate the UMAP plot. Did the integration perform well?
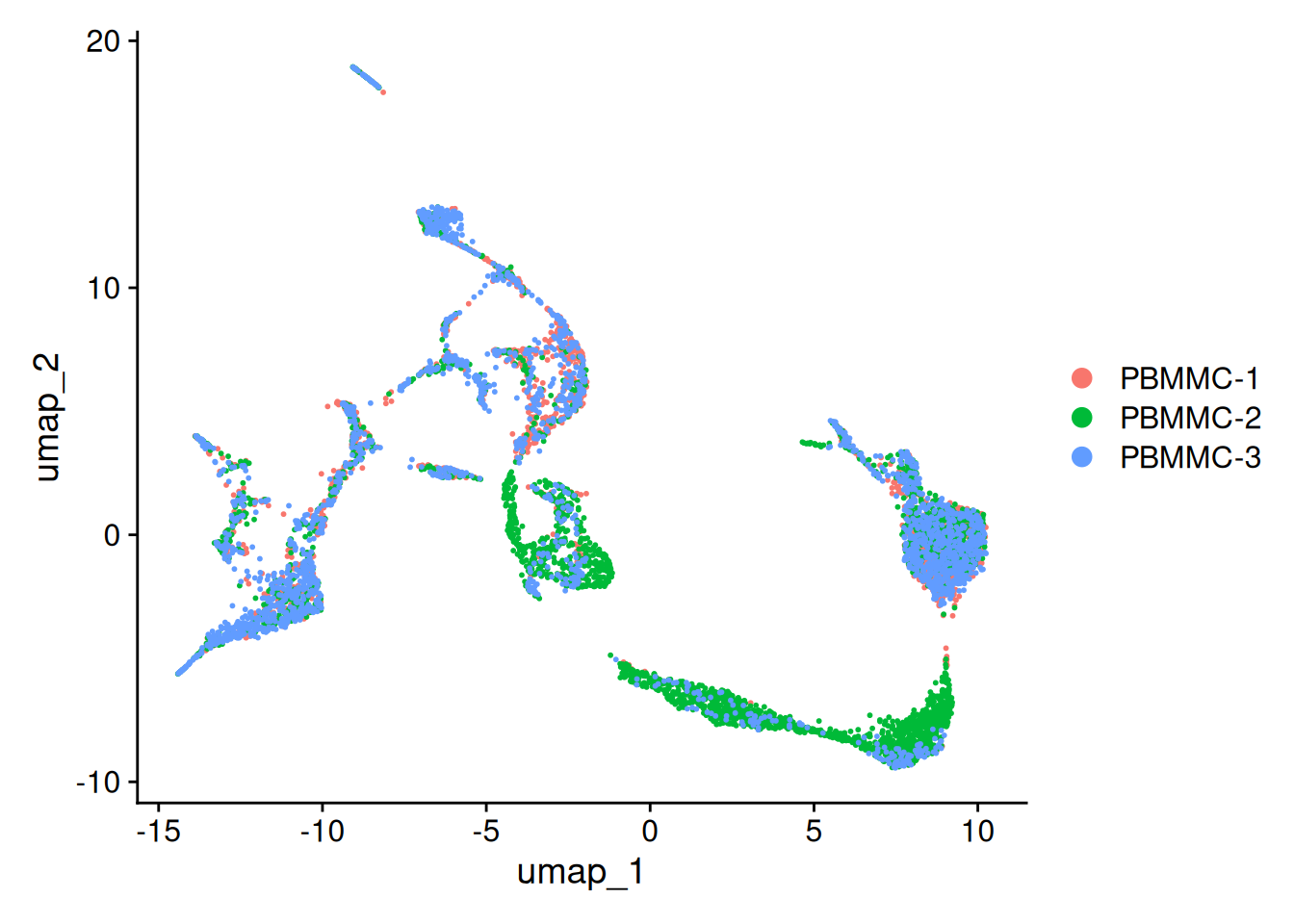
Also in 3D
seu <- RunUMAP(seu, dims = 1:25, n.components = 3, , reduction = "integrated.cca", reduction.name = "umap_3D")15:08:25 UMAP embedding parameters a = 0.9922 b = 1.11215:08:25 Read 6830 rows and found 25 numeric columns15:08:25 Using Annoy for neighbor search, n_neighbors = 3015:08:25 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
15:08:25 Writing NN index file to temp file /tmp/RtmpL91C93/file2036731b200
15:08:25 Searching Annoy index using 1 thread, search_k = 3000
15:08:27 Annoy recall = 100%
15:08:28 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
15:08:28 Initializing from normalized Laplacian + noise (using RSpectra)
15:08:29 Commencing optimization for 500 epochs, with 297686 positive edges
15:08:29 Using rng type: pcg
15:08:35 Optimization finishedplot_3d_umap(seu = seu, reduction = "umap_3D", sample_col = "orig.ident")Save the dataset and clear environment
saveRDS(seu, "day2/seu_day2-3.rds")Clear your environment:
gc()