Seurat::DimPlot(seu, reduction = "umap")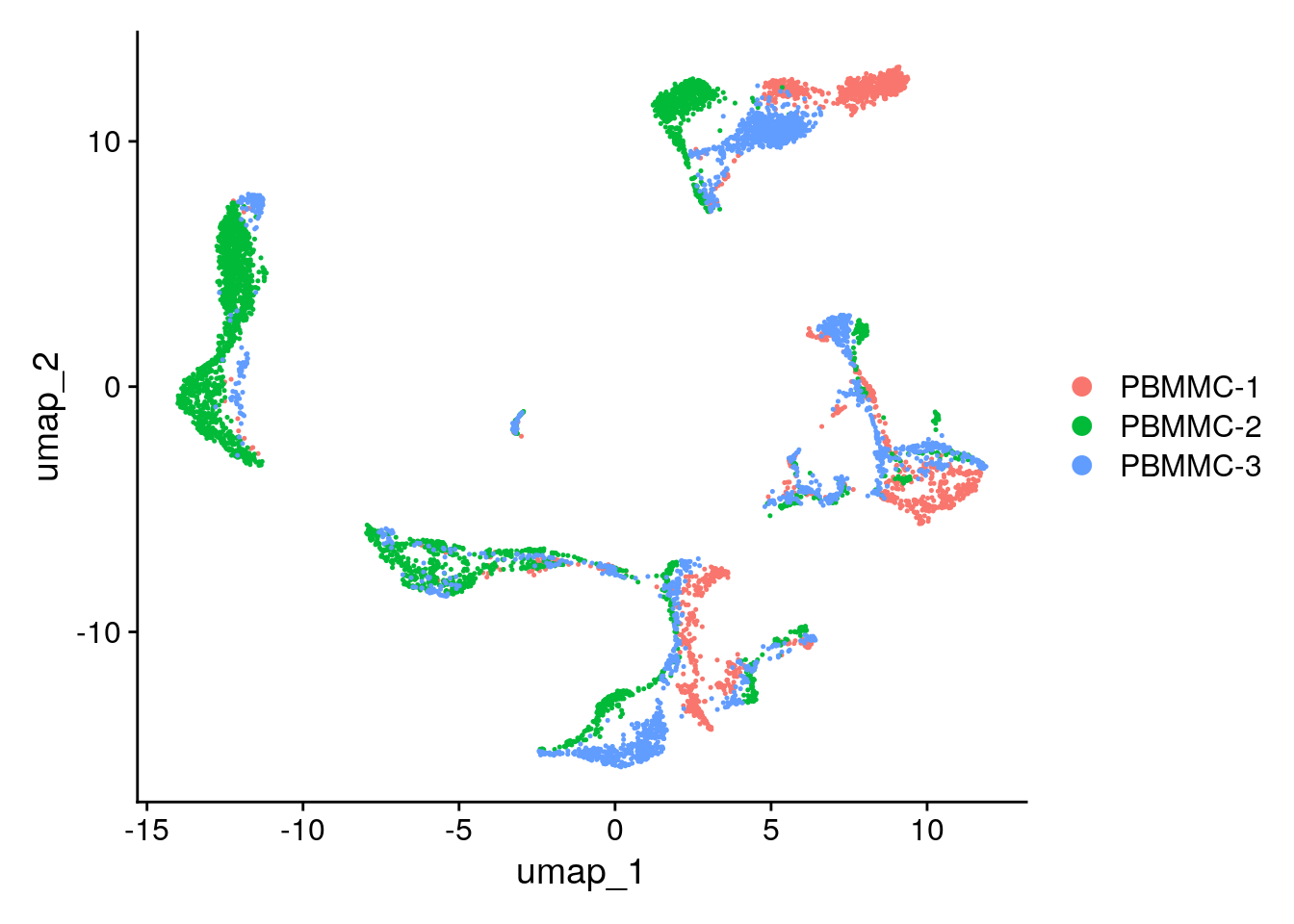
Let’s have a look at the UMAP again. Although cells of different samples are shared amongst ‘clusters’, you can still see seperation within the clusters:
Seurat::DimPlot(seu, reduction = "umap")
To perform the integration, we split our object by sample, resulting into a set of layers within the RNA assay. The layers are integrated and stored in the reduction slot - in our case we call it integrated.cca. Then, we re-join the layers
seu[["RNA"]] <- split(seu[["RNA"]], f = seu$orig.ident)
seu <- Seurat::IntegrateLayers(object = seu, method = CCAIntegration,
orig.reduction = "pca",
new.reduction = "integrated.cca",
verbose = FALSE)
# re-join layers after integration
seu[["RNA"]] <- JoinLayers(seu[["RNA"]])We can then use this new integrated matrix for clustering and visualization. Now, we can re-run and visualize the results with UMAP.
Create the UMAP again on the integrated.cca reduction (using the function RunUMAP - set the option reduction accordingly). After that, generate the UMAP plot. Did the integration perform well?
Performing the scaling, PCA and UMAP:
seu <- RunUMAP(seu, dims = 1:30, reduction = "integrated.cca")Plotting the UMAP:
Seurat::DimPlot(seu, reduction = "umap")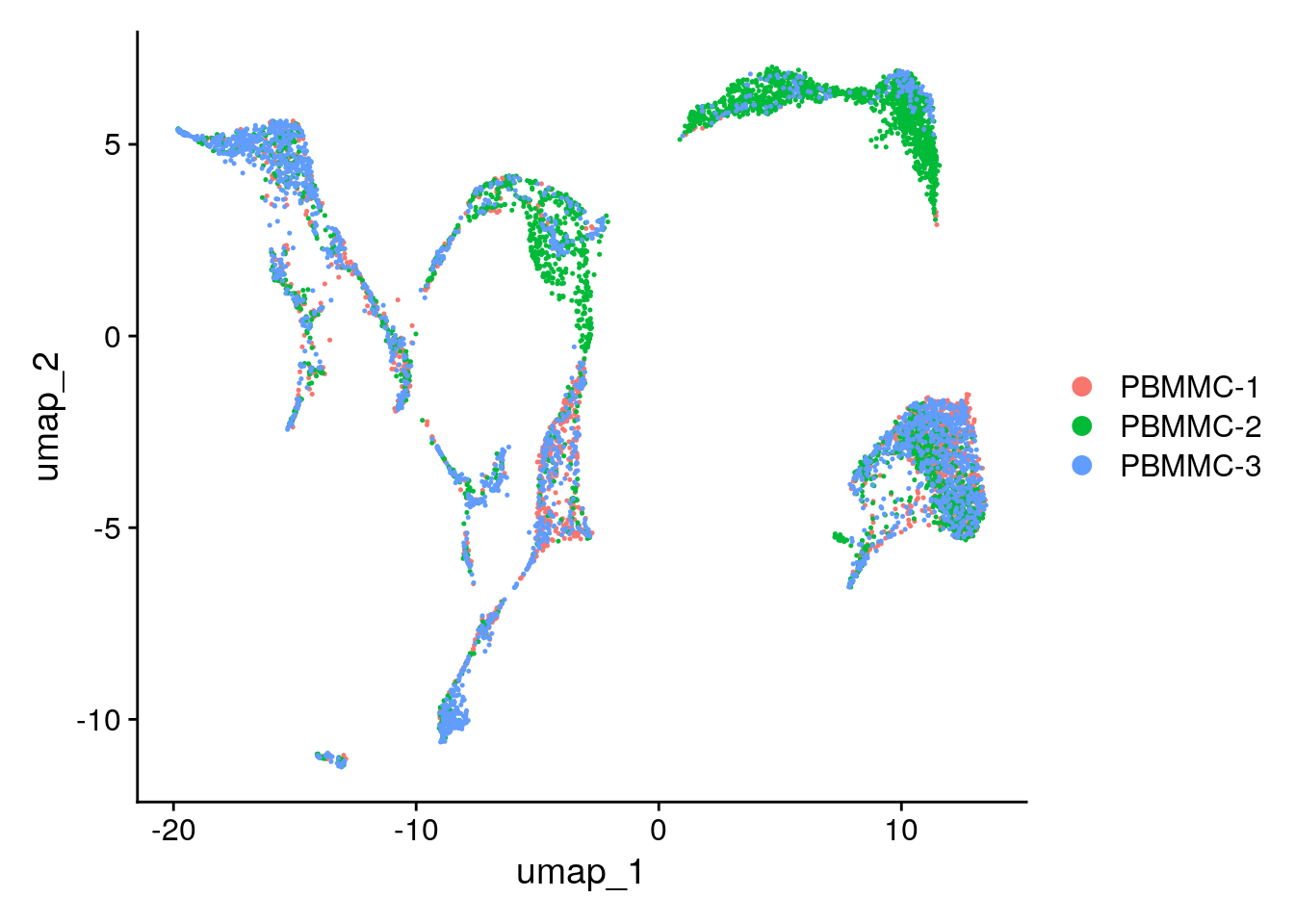
saveRDS(seu, "seu_day2-2.rds")Clear your environment: